用于微卫星不稳定检测的引物组和方法与流程
本发明涉及核酸检测
技术领域:
,具体涉及用于微卫星不稳定检测的引物组和方法。
背景技术:
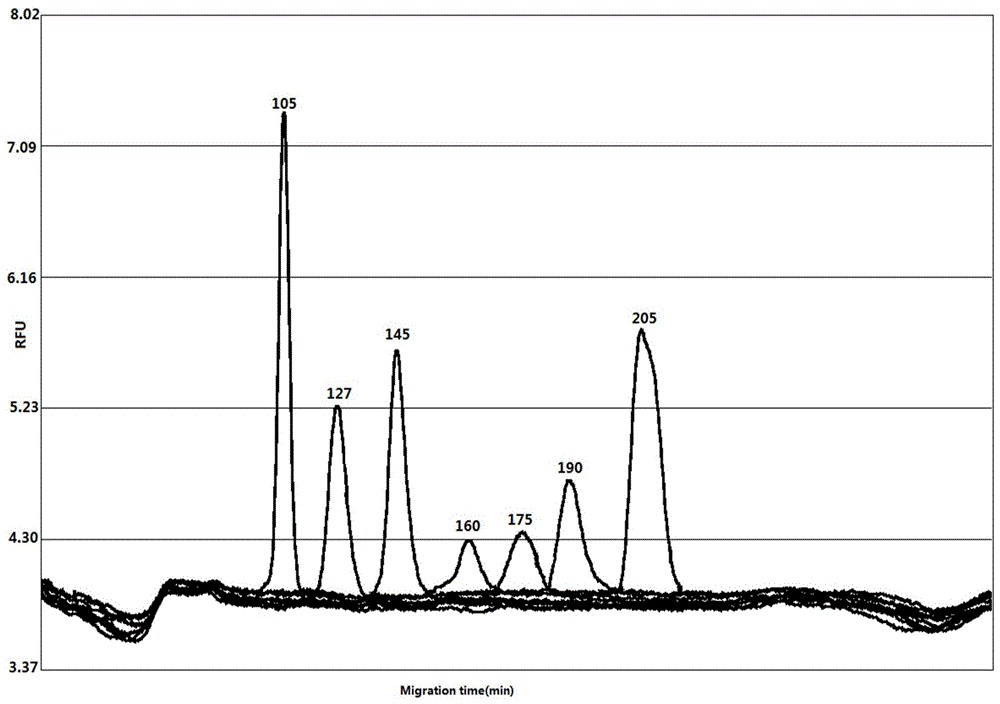
:微卫星(microsatellite,ms)是基因组中小于10个核苷酸的短串联重复序列,一般长度为1-6个核苷酸。微卫星存在于整个基因组中,常见于基因的内含子中,在启动子、转录非翻译区(utr)和外显子中也有分布。在人基因组中,预测至少存在50万个ms位点。微卫星不稳定(microsatelliteinstability,msi)是由于dna错配修复系统(dnamismatchrepairsystem,mmr)出现缺陷而造成的复制错误(replicationerror,rer)引起的简单序列的增加或减少,也就是微卫星长度的增加或减少。因此,msi也称为rer表型。错配修复系统缺陷主要包含两种:(1)错配修复基因mlh1、msh2、msh6和pms2中的一个或者多个发生胚系突变,导致错配修复缺陷;(2)mlh1启动子区域的超甲基化。研究表明,约15%的结直肠癌患者表现为微卫星高不稳定(msi-h),90%以上的遗传性非息肉病性大肠癌(hereditarynon-polyposiscolorectalcancer,hnpcc)患者具有msi-h特征。msi-h现象也会散发在子宫内膜癌、卵巢癌、胃癌等多种癌症中。1993年,altonen首先在hnpcc细胞中发现大量msi现象。1997年,确定了msi的学术名称、定义、检测方法、在crc和其他癌症中的水平等各项相关标准,其中包括5个检测msi的参考marker(bat25,bat26,5s346,d2s123,d17s250,也称为bethesdapanel),该方法采用pcr联合变性电泳检测。由于经实验证明双碱基marker的特异性没有单碱基marker高,因此,在2002年nci会议上,对bethesdapanel重新进行评定,建议采用单碱基和五碱基marker。bacher等人采用荧光pcr联合毛细管电泳的方法筛选了266个msi位点,确定了由7个markers组成的检测panel,包括bat-25、bat-26、nr-21、nr-24和mono-27(msi级别判定)以及pentac和pentad(识别潜在样品混杂和交叉污染)。promega公司开发了包括bat-25、bat-26、nr-21、nr-24和mono-27(msi级别判定)以及amel、pentac和pentad(识别潜在样品混杂和交叉污染)的msi检测试剂盒,并通过了fda批准。msi的pcr检测方法是判断微卫星不稳定的金标准:存在2个及以上微卫星位点不稳定的样本判断为微卫星高不稳定样本(msi-h)、存在1个微卫星位点不稳定的样本判断为微卫星低不稳定样本(msi-l)或微卫星稳定(mss)。研究表明检测患者的msi分型具有重要临床意义。ii期结直肠癌患者中,msi-h表型的患者预后良好;但是,msi-h表型的患者不能从5-氟尿嘧啶治疗中获益。用于微卫星不稳定检测的样本类型通常包括新鲜组织和ffpe样本。其中,ffpe样本的dna降解严重,故msi的pcr产物不能太长,否则会导致扩增失败、无扩增产物。为实现对降解严重的ffpe样本进行有效的检出,一般设计msi的pcr产物大小为80~200bp。此外,在一个pcr反应体系中实现多个微卫星位点的检测,pcr产物很容易出现相互重叠的情况,造成位点的错误判断。现有技术多通过对pcr产物进行分组,使大小相近的pcr产物分到不同的组中,并采用不同的荧光标记的方式对pcr产物进行区分,再利用毛细管电泳检测不同荧光通道的片段大小,得到各个荧光通道的检测结果。然而,该方法存在以下问题:(1)荧光标记成本较高;(2)得到的pcr产物需要依赖于一代测序仪进行检测,大大增加了检测的人力、物料和时间成本以及方法的繁琐性,无法广泛推广和应用;并且使用一代测序仪时,需要用到甲酰胺进行dna变性,而甲酰胺是高毒性物质,对操作人员的健康有潜在的影响;(3)不同荧光通道因光栅问题导致相互渗透,产生的较高背景值和非特异峰,干扰结果判读。因此,开发一种无需荧光标记、利用普通pcr、在一个反应体系中实现多个msi位点高效检测的方法具有重要意义。技术实现要素:为了解决现有技术中存在的技术问题,本发明的目的在于提供用于微卫星不稳定的多重pcr检测引物组以及含有该引物组的试剂盒。尽管利用不同荧光标记的引物进行msi的检测存在诸多缺陷,但是,若要实现不依赖于荧光标记、在一个pcr反应体系中同时检测多个ms位点(尤其是5个以上的位点),需要很好地克服pcr扩增产物的区分检测问题以及各位点pcr扩增的相互影响(各引物之间以及引物与靶序列之间)等问题。尤其是,受限于ms位点的pcr扩增产物的长度,若要在较短的长度范围内有效区分不同的扩增产物更为困难。本发明通过在ms位点扩增引物的5’端添加0~25个碱基,使得各扩增产物之间相差15bp以上,达到非荧光标记的dna区分检测技术的要求。在上述设计构思的基础上,本发明开发了针对bat25、nr24、mono27、bat26、nr21、nr-27位点以及个体识别位点amel的肿瘤细胞msi检测引物组,该引物组在不使用荧光标记的情况下,实现了在一个pcr反应体系中同时扩增6个ms位点和1个个体识别位点、使用非荧光标记的检测技术(如毛细管电泳)即可实现各pcr扩增产物的高效检测。具体地,本发明的技术方案如下:本发明提供一种微卫星不稳定的多重pcr检测引物组,所述引物组中的引物包含检测靶标互补配对区和检测靶标非互补配对区;所述检测靶标非互补配对区位于所述引物的5’端,长度为0~25bp;通过调整所述引物组中各引物所述检测靶标非互补配对区的长度,使得各引物的目的扩增产物之间长度差异至少为15bp。本发明中,所述检测靶标互补配对区为能够与检测靶标互补配对的序列;所述检测靶标非互补配对区为无法与检测靶标序列互补配对的序列。所述通过调整各引物中所述检测靶标非互补配对区的长度,使得各引物的扩增产物之间长度差异至少为15bp具体为:根据各引物对的所述检测靶标互补配对区确定各扩增产物的长度,再综合所述引物组中各引物的扩增产物长度确定在其对应的引物5’端(在正向引物和/或反向引物的5’端)添加碱基的数目,在满足添加碱基的数目为0~25个的情况下,使得各扩增产物之间的长度差异至少为15bp。为保证引物的扩增效率,在5’端添加的序列应尽量不影响引物与靶序列的结合和扩增,同时,应尽量降低非特异性扩增的发生。本发明中,所述引物组中的引物无荧光标记修饰。在一个反应体系中能够同时扩增的ms位点数目受到扩增产物长度的限制。具体地,当所述扩增产物的长度为80~200bp时,所述引物组包含的用于扩增微卫星不稳定标记的引物对不超过10个。本发明提供一种微卫星不稳定检测试剂盒,其包含所述微卫星不稳定的多重pcr检测引物组。进一步地,基于上述微卫星不稳定的多重pcr检测引物组的设计构思,针对特定的ms位点,通过优化5’端检测靶标非互补配对区的碱基组成以及检测靶标互补配对区的序列,使得两者能够较好地适配,在不影响引物特异性扩增效率同时降低非特异性扩增,同时尽量降低各引物对之间相互作用对于扩增造成的不利影响,本发明提供了用于肿瘤细胞微卫星不稳定的多重pcr检测的引物组,所述引物组包括分别用于扩增微卫星位点bat25、nr24、mono27、bat26和nr21的引物对,序列如seqidno.1-10所示。引物序列具体如下:seqidno.1:bat25-f:gtcctcgcctccaagaatgtaag;seqidno.2:bat25-r:gtctgcattttaactatggctc;seqidno.3:nr24-f:gtgtcttgctgaattttacctcctga;seqidno.4:nr24-r:gcagtgagcggagattgtgcc;seqidno.5:nr27-f:cctggaaacaaagcattgaagtctg;seqidno.6:nr27-r:gaggttctgagtcgattaatactag;seqidno.7:mono27-f:tcggtggatcaaatttcacttgg;seqidno.8:mono27-r:tgaaatggtgggaacccag;seqidno.9:bat26-f:ttccctttggacagtttgaactgac;seqidno.10:bat26-r:agctcctttataagcttcttcagt。在上述5对引物的基础上,还可进一步引入其他ms扩增引物对。优选地,所述引物组还包括用于扩增微卫星位点nr27的引物对,所述引物对的序列如seqidno.11-12所示。上述引物对序列经优化设计,其引入仍能够保证扩增体系中各ms位点的扩增效率同时降低非特异性扩增。seqidno.11:nr21-f:cgtctgagaccaagcagataaaagagaacacg;seqidno.12:nr21-r:cggatgtgtaccctttcagcagaattccagcc。在上述引物组中还需进一步引入个体识别位点,以识别潜在样品混杂和交叉污染。优选地,所述引物组还包括用于扩增个体识别位点ame1的引物对,所述引物对的序列如seqidno.13-14所示。seqidno.13:ame1-f:ccctgggctctgtaaagaatag;seqidno.14:ame1-r:atcagagcttaaactgggaagctg。进一步地,本发明还提供所述肿瘤细胞微卫星不稳定的多重pcr检测引物组在制备用于检测肿瘤细胞微卫星不稳定的试剂或试剂盒中的应用。本发明提供一种肿瘤细胞微卫星不稳定检测试剂盒,其包括所述肿瘤细胞微卫星不稳定的多重pcr检测引物组。优选地,所述试剂盒在工作时,所述肿瘤细胞微卫星不稳定的多重pcr检测引物组的各引物在pcr反应体系中的终浓度为bat25扩增引物对0.1~0.5μm,nr24扩增引物对0.2~1μm,mono27扩增引物对0.2~2μm,bat26扩增引物对0.1~3μm,nr21扩增引物对0.1~1μm,nr27扩增引物对0.1~2μm,amel扩增引物对0.01~0.1μm。更优选地,各引物浓度为:bat25扩增引物对0.1μm,nr24扩增引物对0.2μm,mono27扩增引物对2μm,bat26扩增引物对1μm,nr21扩增引物对0.3μm,nr27扩增引物对1μm,amel扩增引物对0.05μm。本发明还提供一种微卫星不稳定检测方法,包括:利用所述微卫星不稳定的多重pcr检测引物组或所述肿瘤细胞微卫星不稳定的多重pcr检测引物组进行多重pcr扩增,检测pcr扩增产物的片段大小。具体地,以待测样本的dna为模板,利用所述引物组进行多重pcr扩增,利用dna检测设备对pcr扩增产物的片段大小进行检测。优选地,所述pcr扩增的反应体系中,各引物浓度为:bat25扩增引物对0.1μm,nr24扩增引物对0.2μm,mono27扩增引物对2μm,bat26扩增引物对1μm,nr21扩增引物对0.3μm,nr27扩增引物对1μm,amel扩增引物对0.05μm。更优选地,所述多重pcr扩增的20μl反应体系如下:5×酶混合液4μl,引物混合物1μl(各引物在pcr反应体系的终浓度为bat25扩增引物对0.1μm,nr24扩增引物对0.2μm,mono27扩增引物对2μm,bat26扩增引物对1μm,nr21扩增引物对0.3μm,nr27扩增引物对1μm,amel扩增引物对0.05μm),dna模板2μl,水13μl。优选地,所述多重pcr扩增的反应程序包括:95℃10min;95℃5min;94℃30sec,60℃30sec,70℃30sec,30个循环;70℃5min。利用本发明所述的引物组可实现利用非荧光标记检测设备即可区分检测各ms位点扩增产物。用于ms位点扩增产物的检测设备包括但不限于qsep100全自动核酸分析仪、labchip、analyzer2100等。本发明的有益效果在于:本发明的微卫星不稳定检测引物组和检测方法具有如下优势:无需荧光标记,即可实现在一个pcr反应体系中同时检测6个微卫星位点,成本明显降低且操作步骤简单;检测不依赖于一代测序仪,不需要使用有毒物质甲酰胺,安全性更高且通量更灵活;避免了荧光相关干扰导致的背景值和非特异峰问题,扩增效率高、特异性高,检测结果更加准确可靠。本发明的引物组和检测方法既可适用于新鲜样本,也可适用于降解严重的ffpe样本,具有广泛的应用前景。附图说明图1为本发明实施例3中利用qsep100全自动核酸分析仪对m2患者的对照样本(外周血)的ms位点pcr扩增产物进行检测的结果图。图2为本发明实施例3中利用qsep100全自动核酸分析仪对m2患者的肿瘤组织样本的ms位点pcr扩增产物进行检测的结果图。具体实施方式下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。实施例1用于微卫星不稳定检测的引物组的设计针对ms位点bat25、nr24、mono27、bat26、nr21和nr27以及个体识别位点amel设计扩增引物,通过在各引物对的正向引物和反向引物的5’端添加0~25bp范围内不同长度的序列,使得各位点的目的扩增产物之间的长度(扩增产物的理论长度)差异至少为15bp。引物设计和筛选的整体流程如下:第一步,根据人基因组序列信息,针对ms位点bat25、nr24、mono27、bat26、nr21和nr27以及个体识别位点amel进行pcr引物设计,目标产物大小为80-200bp。经筛选获得能够保证每个检测位点的扩增效率的特异性较高、退火温度相近的引物。第二步,计算第一步筛选得到的各位点引物的预期扩增产物大小,对产物大小间隔小于15bp的引物,进行二次设计。对该引物对的正向引物和/或反向引物的5’端添加靶标非互补序列,使产物大小与其他产物大小相差至少15bp。靶标非互补序列应在人基因组上无特异识别区,可以是人工序列,也可以是其他物种序列。第三步,对所有引物序列进行整体分析、筛选和优化,确保引物之间无相互影响,且扩增效率相近。通过对5’端添加序列和靶点结合序列的大量筛选、组合和优化,得到能够保证在一个pcr反应体系中同时高效特异地扩增ms位点bat25、nr24、mono27、bat26、nr21和nr27以及个体识别位点amel的7对引物,引物序列如表1所示。利用表1中的引物进行扩增,ms位点bat25、nr24、mono27、bat26、nr21和nr27的扩增产物长度分别为127bp、145bp、175bp、190bp、205bp和160bp;个体识别位点ame1的扩增产物长度为105bp。表16个ms位点以及1个个体识别位点的扩增引物序列引物名称序列bat25-fgtcctcgcctccaagaatgtaagbat25-rgtctgcattttaactatggctcnr24-fgtgtcttgctgaattttacctcctganr24-rgcagtgagcggagattgtgccnr27-fcctggaaacaaagcattgaagtctgnr27-rgaggttctgagtcgattaatactagmono27-ftcggtggatcaaatttcacttggmono27-rtgaaatggtgggaacccagbat26-fttccctttggacagtttgaactgacbat26-ragctcctttataagcttcttcagtnr21-fcgtctgagaccaagcagataaaagagaacacgnr21-rcggatgtgtaccctttcagcagaattccagccame1-fccctgggctctgtaaagaatagame1-ratcagagcttaaactgggaagctg注:下划线标出的碱基为5’端添加的非互补配对序列。实施例2利用微卫星不稳定检测引物组的检测方法利用实施例1的微卫星不稳定检测引物组进行pcr扩增的反应体系如表2所示,其中引物混合物的组成如下:bat25-f0.1μm、bat25-r0.1μm、nr24-f0.2μm、nr24-r0.2μm、nr27-f1μm、nr27-r1μm、mono27-f2μm、mono27-r2μm、bat26-f1μm、bat26-r1μm、nr21-f0.3μm、nr21-r0.3μm、amel-f0.05μm、amel-r0.05μm。表2pcr反应体系在pcr管里按表2的反应体系组成配置反应液,用枪轻柔地上下吹吸混匀。将装有反应体系的pcr管放入pcr仪,按表3设置程序进行反应。表3pcr反应程序pcr扩增结束后,采用qsep100全自动核酸分析仪进行pcr扩增产物的片段大小分析。实施例3肿瘤组织样本的微卫星不稳定检测利用实施例1的微卫星不稳定检测引物组和实施例2的检测方法对2例肿瘤患者(m1患者,m2患者)的肿瘤组织样本的微卫星不稳定状态进行检测,具体如下:1、dna提取:采用qiagenffpednakit结直肠癌肿瘤组织样本的dna,以外周血作为对照样本,采用天根血液提取试剂盒提取外周血的dna。采用qubit检测提取dna的浓度,并稀释到2ng/μl。2、pcr扩增:利用实施例1的微卫星不稳定检测引物组和实施例2的pcr扩增体系和程序进行pcr扩增。3、pcr扩增产物的检测:按照仪器操作说明,采用仪器配套的稀释液对步骤2得到的pcr产物进行10倍稀释后,取10μl用于qsep100全自动核酸分析仪检测。仪器设置:选用配套的高分辨率卡夹s1,设置检测条件:6kv,分离时间8min。4、结果分析分别对对照样本(外周血)和肿瘤组织样本的微卫星位点进行检测,m1患者和m2患者的qsep100检测的统计结果分别如表4和表5所示。m2患者的对照样本和肿瘤组织样本qsep100检测峰图如图1和图2所示。结果表明,m1患者肿瘤组织和对照在6个ms位点均为单一峰(单一产物),即没有发生微卫星不稳定;m2患者的肿瘤组织在bat25和mono27两个位点各有两个产物峰,且都是出现了比对照小的峰,说明这两个位点的微卫星发生了缺失突变,即微卫星不稳定。根据微卫星不稳定判断规则(存在2个及以上位点不稳定,判断为微卫星不稳定样本),m1样本判断为微卫星稳定样本,m2样本判断为微卫星不稳定样本。表4m1患者的ms位点pcr扩增产物片段大小检测结果表5m2患者的ms位点pcr扩增产物片段大小检测结果以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。序列表<110>北京优迅医学检验实验室有限公司<120>用于微卫星不稳定检测的引物组和方法<130>khp191117163.0<160>14<170>siposequencelisting1.0<210>1<211>23<212>dna<213>人工序列(artificialsequence)<400>1gtcctcgcctccaagaatgtaag23<210>2<211>22<212>dna<213>人工序列(artificialsequence)<400>2gtctgcattttaactatggctc22<210>3<211>26<212>dna<213>人工序列(artificialsequence)<400>3gtgtcttgctgaattttacctcctga26<210>4<211>21<212>dna<213>人工序列(artificialsequence)<400>4gcagtgagcggagattgtgcc21<210>5<211>25<212>dna<213>人工序列(artificialsequence)<400>5cctggaaacaaagcattgaagtctg25<210>6<211>25<212>dna<213>人工序列(artificialsequence)<400>6gaggttctgagtcgattaatactag25<210>7<211>23<212>dna<213>人工序列(artificialsequence)<400>7tcggtggatcaaatttcacttgg23<210>8<211>19<212>dna<213>人工序列(artificialsequence)<400>8tgaaatggtgggaacccag19<210>9<211>25<212>dna<213>人工序列(artificialsequence)<400>9ttccctttggacagtttgaactgac25<210>10<211>24<212>dna<213>人工序列(artificialsequence)<400>10agctcctttataagcttcttcagt24<210>11<211>32<212>dna<213>人工序列(artificialsequence)<400>11cgtctgagaccaagcagataaaagagaacacg32<210>12<211>32<212>dna<213>人工序列(artificialsequence)<400>12cggatgtgtaccctttcagcagaattccagcc32<210>13<211>22<212>dna<213>人工序列(artificialsequence)<400>13ccctgggctctgtaaagaatag22<210>14<211>24<212>dna<213>人工序列(artificialsequence)<400>14atcagagcttaaactgggaagctg24当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1