一种苯并呋喃酮的合成方法与流程
本发明涉及医药中间体制备的技术领域,尤其涉及一种苯并呋喃酮的合成方法。
背景技术:
嘧菌酯是农药界一类极具影响力的新型农用杀菌剂,具有广谱的杀菌活性,对几乎全部的真菌纲病菌害都有良好的活性,并且具有极好的保护、治疗、铲除、渗透、内吸活性等特点。在推荐剂量下使用嘧菌酯不仅可以保护作物的生长,使农作物安全无药害,而且对地下水和环境没有危害。目前,国内生产嘧菌酯类的农药主要从国外进口,由于受技术限制国内生产厂家较少,我国迫切需要新技术填补材料缺乏的空缺。
制备嘧菌酯困难最主要的原因是中间体苯并呋喃酮的合成没有取得突破进展。苯并呋喃酮作为重要的有机合成中间体,广泛存在的一类内酯化合物,具有很强生物活性的化合物,在农药、医药及新型抗氧剂等方面一直是人们研究的热点。
目前,制备苯并呋喃酮的方法主要有三种:
1、以邻硝基甲苯为原料先制得邻硝基苯乙酸,再还原经重氮化反应、水解得到苯并呋喃酮,参照下述合成路线:
但是这种方法步骤繁琐,合成条件苛刻,产率较低。
2、以苯甲醚为原料制得邻羟基苯甲酸再环化生成苯并呋喃酮,但在这个过程中邻羟基苯甲酸制备过程复杂,产率很低,得不到有效的推广;
3、以邻氯苯乙酸为原料经水解环合制备,但是邻氯苯乙酸生产成本较高,不易获得。
苯并呋喃酮因为自身优良的性质得到了广泛的应用,因此其生产规模不断扩大,但目前苯并呋喃酮的合成收率普遍偏低且条件苛刻、操作繁琐,不仅造成大量原料和能量浪费,而且增加了环境污染压力,迫切需要改进制备工艺,寻求苯并呋喃酮高收率和易操作的有效途径。
技术实现要素:
基于背景技术存在的技术问题,本发明提出了一种苯并呋喃酮的合成方法,通过以相对易得的邻甲酚作为起始原料,经四步反应制得目标产物苯并呋喃酮,整个制备过程条件温和,收率高,易操作,且环境污染小,适合大规模工业化生产。
本发明是通过如下技术方案实现的:
一种苯并呋喃酮的合成方法,包括如下步骤:
(1)将邻甲酚进行羟基保护反应,生成中间体(ⅰ);
(2)将中间体(ⅰ)进行甲基羰基化反应,生成含苯乙酸酯结构的中间体(ⅱ);
(3)将中间体(ⅱ)进行酯的水解和羟基脱保护反应,生成含邻羟基苯乙酸结构的中间体(ⅲ);
(4)将中间体(ⅲ)再进行内酯化反应,生成苯并呋喃酮。
优选地,中间体(ⅰ)的结构式为:
r1为羟基保护基,优选为烷基、酰基、硅基、烷氧基烷基、碳酸酯基,进一步优选为甲基、乙基、丙基、异丙基、乙酰基、丙酰基、苯甲酰基、苯磺酰基、对甲苯磺酰基、甲磺酰基、三甲基硅基、三乙基硅基、二甲基异丙基硅基或甲氧基甲基;
为了避免邻甲酚的羟基后续发生氧化、酰基化、卤化、脱水或其他许多官能团的转化等反应时造成影响,本发明在步骤(1)中先将羟基保护起来,具体的可选择在自以往就已知的条件下进行。
例如,烷基醚保护。烷基醚的保护有甲基醚、烯丙基醚、叔丁醚、苄基醚、对甲氧基苄基醚、三苯甲基醚等,在目前使用也比较普遍。一般烷基上的羟基在用苄基醚保护时需要用强碱,但酚羟基的苄基醚保护一般只要用碳酸钾在乙腈或丙酮中回流即可。
酰基化保护。有乙酰基、丙酰基、苯甲酰基、特戊酰基等进行酰化保护,在温和条件下就可以达到很好的保护。
硅醚保护基。硅醚是最常见的保护羟基的方法之一。随着硅原子上的取代基的不同,保护和去保护的反应活性均有较大的变化,空间效应及电子效应是影响反应的主要因素。在进行选择性去保护反应时,硅原子周围的空间效应,以及被保护分子的结构环境均需考虑。tms、tes、tbs、tips、tbdms、tbdps等都可以与羟基形成醚键,在一定条件下对羟基进行保护。
烷氧基烷基醚保护。可以用mom、mtm、mem、bom、sem、thp,烷氧基烷基醚的保护保护方法简单,保护产率较高。
优选地,步骤(1)中,当r1为乙酰基时,将邻甲酚和醋酸酐,在甲苯溶剂下,进行酚羟基乙酰化反应,得到邻乙酰氧基甲苯;
优选地,中间体(ⅱ)的结构式为:
r2为甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、异戊基或环己基;
优选地,步骤(2)中,将中间体(ⅰ)和烷基醇、氧化剂、一氧化碳,在以过渡金属催化剂前体与配体形成的络合物为催化剂的条件下,进行甲基羰基化反应,得到含苯乙酸酯结构的中间体(ⅱ)。
优选地,所述氧化剂为双氧水、过氧乙酸、过氧叔丁醇、氧气、过硫酸钾、间氯过氧苯甲酸中的一种或者多种的组合;
所述过渡金属催化剂前体为钯系金属催化剂前体、钌系金属催化剂前体、铑系金属催化剂前体、钴系金属催化剂前体、镍系金属催化剂前体或铜系金属催化剂前体;
钯系金属催化剂前体优选为氯化钯、钯碳、四三苯基磷钯、二氯化二(三苯基磷)钯、醋酸钯、三氟乙酸钯、三氟甲磺酸钯中的一种或者多种的组合,钌系金属催化剂前体优选为三氯化钌、十二羰基三钌中的一种或者多种的组合,铑系金属催化剂前体优选为醋酸铑、十二羰基四铑、乙烯基氯化铑中的一种或者多种的组合,钴系金属催化剂前体优选为羰基钴、氯化钴、乙酰丙酮钴中的一种或者多种的组合,镍系金属催化剂前体优选为溴化镍、醋酸镍、硫酸镍、氯化镍中的一种或者多种的组合,铜系金属催化剂前体优选为氟化铜、氯化铜、乙酰丙酮铜中的一种或者多种的组合;
所述配体为膦配体和/或氮配体,膦配体优选为1,3-双(二苯基膦)丙烷(dppp)、1,1'-双(二苯基膦)二茂铁(dppf)、1,2-双(二苯膦)乙烷(dppe)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(xantphos)、2,2’-双-(二苯膦基)-1,1’-联萘(binap)、三苯基膦(pph3)中的一种或者多种的组合,氮配体优选为吡啶、2,2’-联吡啶、n,n`-二苯基对苯二胺(dppd)、2,2’-联喹啉和1,10-菲啰啉中的一种或者多种的组合。
优选地,步骤(2)中,将中间体(ⅰ)和烷基醇、氧化剂、络合物混合后,在压力为1-50atm的一氧化碳条件下,升温至80-150℃反应,得到中间体(ⅱ)。
优选地,中间体(ⅲ)的结构式为:
优选地,步骤(3)中,将中间体(ⅱ)先进行酯的水解反应,再进行羟基脱保护反应,得到中间体(ⅲ),或者将中间体(ⅱ)先进行羟基脱保护反应,再进行酯的水解反应,得到中间体(ⅲ);或者将中间体(ⅱ)同时进行酯的水解反应和羟基脱保护反应,得到中间体(ⅲ)。
优选地,进行酯的水解反应时,在碱性条件下先进行酯化水解反应,再在酸性条件下进行酸化,如此将中间体(ⅱ)中的苯乙酸酯结构转化成苯乙酸结构;
进行羟基脱保护反应时,当中间体(ⅱ)中的r1为甲基时,在路易斯酸催化剂的条件下,进行羟基脱保护反应;当中间体(ⅱ)中的r1为乙酰基时,分别在碱性和酸性条件下进行酯的水解反应的同时也进行了羟基脱保护反应。
优选地,步骤(4)中,将中间体(ⅲ)在酸催化剂的条件下,进行内酯化反应,得到苯并呋喃酮;
优选地,步骤(4)中,将中间体(ⅲ)加入带水剂中,再加入酸催化剂进行反应,分别脱除水和有机溶剂,即得苯并呋喃酮。
反应过程中,反应生成的水与带水剂形成共沸物一起经冷凝后进入分水器,可以促进酯化反应不断地向正反应方向进行,在分水器中进一步形成水层和油层,除去水层,油层回流进入混合体系,反应结束后进一步得到苯并呋喃酮。
优选地,所述酸催化剂为固体磺酸、固体磷酸、硫酸氢钾、磷酸二氢钾、硫酸、乙酸、甲基磺酸和硅胶磺酸中的一种或者多种的组合;
所述带水剂为芳烃、烷烃和/或卤代烃,优选为甲苯、二甲苯、三甲苯、氯苯、己烷中的一种或者多种的组合;
优选地,所述带水剂的用量与中间体(ⅲ)的摩尔比为3-15:1;
优选地,本发明所述苯并呋喃酮的合成方法合成路线如下所示:
与现有技术相比,本发明具有如下优点:
本发明采用非常易得的原材料通过一种环保高效的生产工艺来制备苯并呋喃酮。
首先本发明以邻甲酚作为原材料,邻甲酚可以从煤焦油分馏而得,随着科技的进步又发展了许多合成邻甲酚的方法,有甲苯磺化法、甲基异丙基苯氧化法等,邻甲酚的制备过程简单,原料易得。
之后本发明通过将邻甲酚中的羟基保护起来,与醇、氧化剂和一氧化碳在过渡金属催化剂下通过一步c(sp3)-h直接羰基化得到苯乙酸酯类化合物,再通过碱性水解,脱保护得到邻羟基苯甲酸,最后进行内酯化反应,即得到我们所需要的苯并呋喃酮,苯并呋喃酮可以作为许多材料合成的中间体,大量的用于食品香味剂、农药、医药等行业,在工业生产方面具有很大的发展空间。
和已报道的方法相比,本发明合成方法采用的生产工艺环保无污染,反应条件温和,原料易得,步骤简单,产品收率和纯度都比较高,相比较现有的工艺得到了很大的改善。
附图说明
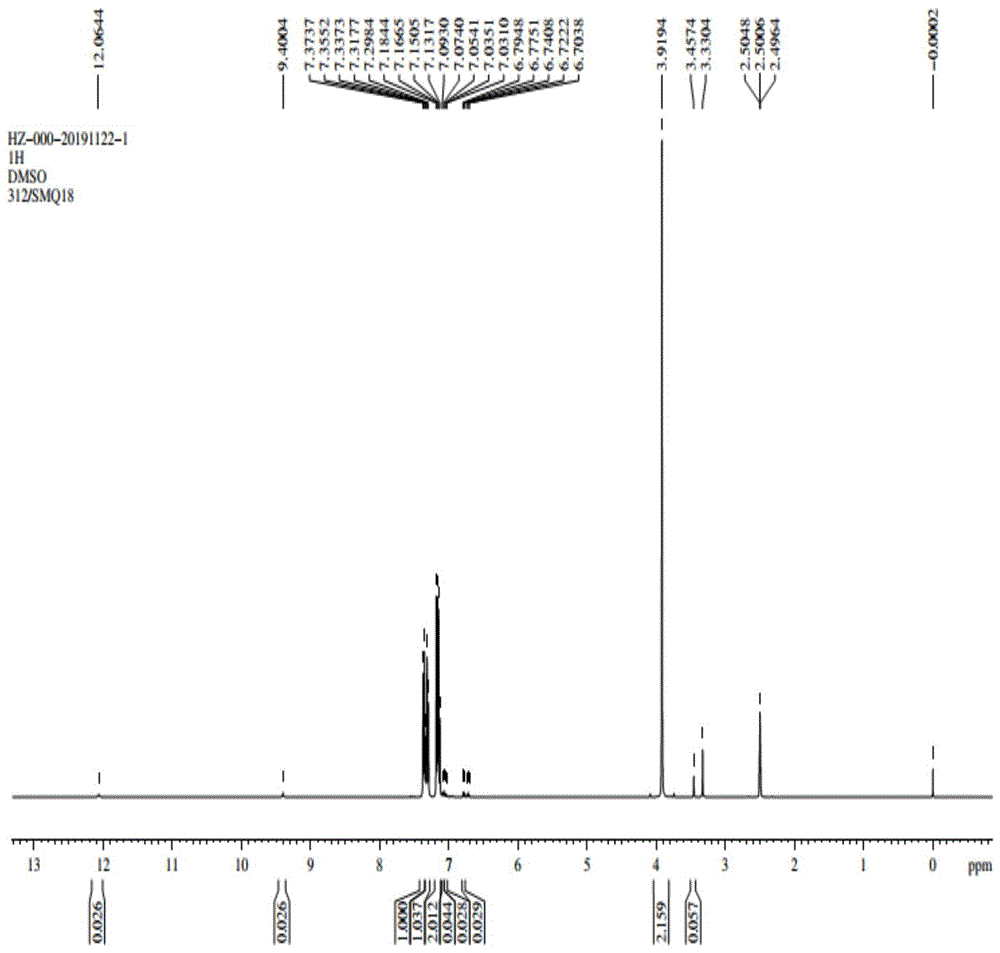
图1为苯并呋喃酮的1hnmr表征图谱;
图2为苯并呋喃酮的13cnmr表征图谱;
图3为苯并呋喃酮的色谱表征图谱。
具体实施方式
实施例1
一种苯并呋喃酮的合成方法,其合成路线如下:
具体包括如下步骤:
(1)、将250ml四口烧瓶放在10℃冰浴中,加入100ml10wt%的naoh溶液,在搅拌下加入邻甲酚10.8g(0.1mol),搅拌5min,然后滴加硫酸二甲酯7.5g(60mmol),滴加完毕后,升温至100℃,保持温度反应5h,冷却至室温,用水冲洗分出油层,水层用二氯甲烷提取,油层和萃取液合并,依次用稀naoh溶液和水洗涤,无水硫酸镁干燥,旋蒸出二氯甲烷,得到邻甲氧基甲苯11.67g,收率95.6%;
(2)、将邻甲氧基甲苯12.2g(100mmol)、乙醇1.84g(40mmol)、过氧叔丁醚5.84g(40mmol)、氟化铜(0.6mg),xantphos(2.9mg),加入100ml反应釜中,通入一氧化碳到50atm,升温到120℃,搅拌反应16小时,期间维持一氧化碳压力在50atm,反应结束后冷却至室温,排出一氧化碳,用亚硫酸氢钠水溶液洗涤,干燥,蒸馏回收原料,得到蒸馏后的有机相备用;
(3)、向步骤(2)得到的蒸馏后的有机相中加入20ml6mol/l的氢氧化钠水溶液,加热到50℃搅拌反应2h,反应结束后冷却至室温,用2mol/l的盐酸调ph到1,析出固体,过滤,水洗干燥,得到邻甲氧基苯乙酸5.4g,收率82%(以乙醇获取的邻甲氧基苯乙酸理论量为基准);
将邻甲氧基苯乙酸16.6g(0.1mol)、二氯乙烷200ml加入到500ml三口烧瓶中,搅拌均匀后加入无水alcl35g(0.037mol),搅拌反应12h,反应结束后加入50ml水,用盐酸酸化,水洗,干燥,浓缩,得到邻羟基苯乙酸,收率89%;
(4)、将邻羟基苯乙酸15.2g(100mmol)、三甲苯100ml加入到250ml三口烧瓶中,装上分水器,开启搅拌器并加热三口烧瓶,加热至150℃时加入0.6g硫酸氢钾,将混合物升温回流反应6h后结束,反应液冷却至室温,过滤除去催化剂,再依次用亚硫酸氢钠和水洗涤滤液,用无水硫酸镁干燥,蒸馏除去三甲苯,即得苯并呋喃酮,收率为97%。
实施例2
一种苯并呋喃酮的合成方法,其合成路线如下:
具体包括如下步骤:
(1)、在250ml四口瓶中,加入邻甲酚10.8g(100mmol)、甲苯100ml,加热至回流,滴加11.2g醋酸酐(110mmol),滴加完后继续回流反应2小时,tlc检测邻羟基甲苯已经转化完全,浓缩脱除溶剂和醋酸,蒸馏得到邻乙酰氧基甲苯14.4g,收率96%;
(2)、将邻乙酰氧基甲苯15g(100mmol)、乙醇1.84g(40mmol),过氧叔丁醚5.84g(40mmol),三氯化钌(1.1mg),xantphos(2.9mg),加入100ml反应釜中,通入一氧化碳到50atm,升温到150℃,搅拌反应10小时,期间维持一氧化碳压力在50atm,反应结束后冷却至室温,排出一氧化碳,亚硫酸氢钠水溶液洗涤,干燥,蒸馏回收原料,得到蒸馏后的有机相备用;
(3)、向步骤(2)得到的有机相中加入20ml6mol/l的氢氧化钠水溶液,加热到50℃搅拌反应2h,反应结束后冷却至室温,用2mol/l的盐酸调ph到1,析出固体,过滤,水洗干燥,得到邻羟基苯乙酸5.2g,收率85%;
(4)、将邻羟基苯乙酸15.2g(100mmol)、甲苯100ml加入到250ml三口烧瓶中,装上分水器,开启搅拌器并加热三口烧瓶,加热至50℃,加入0.5g硅胶磺酸,将混合物升温回流反应6h后结束,反应液冷却至室温后,过滤除去催化剂,再依次用亚硫酸氢钠和水洗涤滤液,用无水硫酸镁干燥,蒸馏除去甲苯,即得苯并呋喃酮,收率为97%。
实施例3
一种苯并呋喃酮的合成方法,其合成路线如下:
具体包括如下步骤
(1)、在250ml四口瓶中,加入邻甲酚10.8g(100mmol)、二氯乙烷100ml、二异丙基乙胺30ml(0.2mol)一起搅拌10min,再加入氯甲基甲醚10ml(ch3och2cl,0.15mol),将混合物在室温下搅拌反应10h,停止反应后加入饱和nh4cl溶液,分离溶液相,将有机相浓缩,蒸馏得到2-(甲氧基甲氧基)甲苯14.9g,收率98%;
(2)、将2-(甲氧基甲氧基)甲苯15.2g(100mmol),乙醇1.84g(40mmol),过氧叔丁醚5.84g(40mmol)、pd(xantphos)cl215.2mg(0.02mmol)加入100ml反应釜中,通入一氧化碳到10atm,升温到80℃,搅拌24小时,期间维持一氧化碳压力在10atm,反应结束后冷却至室温,冷却排出一氧化碳,用亚硫酸氢钠水溶液洗涤,干燥,蒸馏回收原料,得到蒸馏后的有机相备用;
(3)、将步骤(2)得到的有机相中加入20ml6mol/l的氢氧化钠水溶液,加热到50℃搅拌反应2h,反应结束后冷却至室温,在搅拌下加入1ml浓盐酸,将混合物在60℃下搅拌回流反应2h,冷却至室温,析出固体,过滤,水洗干燥,得到邻羟基苯乙酸4.9,收率81%;
(4)、将邻羟基苯乙酸15.2g(100mmol)、甲苯100ml加入到250ml三口烧瓶中,装上分水器,开启搅拌器并加热三口烧瓶,加热至100℃时滴加1ml浓度为8mol/l的稀硫酸,滴加完毕后将混合物升温回流反应6h后结束,反应液冷却至室温后,依次用亚硫酸氢钠和水洗涤滤液,用无水硫酸镁干燥,蒸馏除去甲苯,即得苯并呋喃酮,收率为98%。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明的技术范围内,根据本发明的技术方案及其发明加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!