一种创建矮壮株型菊花的方法与流程
本发明属于植物基因工程和转基因育种领域,涉及一种通过转基因技术创制矮状株型菊花的方法及其应用。
背景技术:
菊花(chrysanthemummorifolium)起源于中国,栽培历史悠久,是我国十大传统名花和世界四大切花之一。菊花具有丰富的花色、多样的花姿等特点,深受人们的喜爱。株型直接影响花卉的观赏品质,是育种者关注的主要性状之一。菊花根据用途不同分可为地被菊、切花菊、盆栽菊、案头菊等。育种工作者通过不断的调整栽培方式来调控株型,使之达到不同的用途。其中案头菊要求植株矮,茎秆健壮挺立,株高一般控制在20厘米以内,无分枝。生产上通过缩短营养生长时间,控制氮肥,摘心,使用生长抑制剂等方法来调控株型,耗费人力物力。因此培育出植株矮小,茎秆粗壮的菊花品种将大大降低菊花生产成本,提高效益,增加菊农收入。
近年来,随着分子生物学的迅速发展,通过农杆菌介导的遗传转化的技术在植物体内过量表达或定点敲除特定的内源基因以改变作物的农艺性状已成为现代育种的重要手段。相比传统的遗传育种,这种育种方式大大缩短了育种时间、提高了育种效率,为改良菊花品种提供了新的手段和途径。在菊花中通过转基因技术来改变株型的研究还不完善,尤其是创制植株矮小,茎秆粗壮的菊花还未见报道。因此通过农杆菌介导法将菊花的yab3.1基因导入切花菊‘神马’调控菊花高度是一种新的探索,为采用基因工程技术选育菊花观赏品种提供新颖而实用的方法。
技术实现要素:
本发明的目的在于解决定向改良菊花株型的问题,提供一种cmyab3.1基因,该基因的序列为seqidno.1。
本发明的第二个目的是提供一种cmyab3.1基因植物表达载体。
本发明的另一目的在于提供一种通过转cmyab3.1基因调控菊花株型的方法。本发明涉及的农杆菌介导遗传转化,转基因植物的分子检测及转基因植株后代表型观察用于本发明,为利用基因工程技术开展观赏性菊花分子育种提供方法和实例。
本发明的又一目的在于提供cmyab3.1基因、cmyab3.1基因植物表达载体、调控菊花株型的方法在调控菊花株型方面的基因工程应用。
本发明的目的可以通过以下技术方案实现:
本发明的第一个目的是提供一种改变菊花株型的基因cmyab3.1,该基因的核苷酸序列如seqidno.1所示。
本发明的第二个目的是提供一种植物表达载体,所述植物表达载体包含上述基因cmyab3.1。
进一步的,所述植物表达载体是将上述基因cmyab3.1克隆到过表达载体pore-r4-35saa中所得。
本发明的第三个目的是提供一种调控菊花株型的方法,所述方法包括以下步骤:
(1)菊花cmyab3.1的克隆,获得基因cmyab3.1;
(2)植物表达载体pore-r4-35saa-cmyab3.1的构建:
根据cmyab3.1基因全长序列设计引物,在cmyab3.1基因的上游和下游引物中分别引入xhoi和smai酶切位点,用高保真酶进行pcr反应,然后用限制性内切酶xhoi和smai对pcr产物和植物过表达载体pore-r4-35saa进行双酶切;酶切产物分别回收,再将其回收产物用连接酶连接后,转化dh5α感受态细胞,提取阳性质粒,植物表达载体pore-r4-35saa-cmyab3.1构建成功;
(3)农杆菌eha105介导叶盘法将步骤(2)构建的cmyab3.1基因植物表达载体转入菊花‘神马’中,进行培养初步获得抗性植株:
将步骤(2)获得的植物表达载体pore-r4-35saa-cmyab3.1转化农杆菌感受态细胞eha105,获得阳性克隆农杆菌,通过叶盘法将基因cmyab3.1导入菊花,经过卡那霉素抗性筛选得到阳性转化植株,对阳性转化植株进行pcr、荧光定量rt-pcr检测验证cmyab3.1基因已经插入转基因植株的基因组dna中并表达,获得阳性转基因菊花株系。
进一步的,步骤(1)具体操作为:
以切花菊‘神马’的cdna为模板,根据序列信息设计cmyab3.1基因正、反向特异引物,以反转录的cdna为模板,进行pcr扩增,产物连接到pmd19-t载体,转化dh5α感受态细胞;或根据seqidno.1所示基因序列直接合成,获得seqidno.1所示目的基因的全长序列;
所述cmyab3.1基因正、反向特异引物为:
上游引物cmyab3.1-f:5′-atgtctaccatcacagccac-3′(seqidno.2),
下游引物cmyab3.1-r:5′-ctaataataaggagagacaccaac-3′(seqidno.3);
进一步的,步骤(2)中所述引物为:
上游引物cmyab3.1-xhoi-f:5′-ccgctcgagatgtctaccatcaca-3′(seqidno.4),
下游引物cmyab3.1-smai-r:5′-tcccccgggataataaggagagac-3′(seqidno.5)。
进一步的,步骤(3)所述的对阳性转化植株进行pcr、定量rt-pcr检测的过程为:
1)pcr检测:
取生根筛选得到的卡那霉素抗性植株嫩叶及未转化株嫩叶,提取基因组dna,将gfp的一段序列和cmyab3.1的一段序列作为检测目标,在gfp和cmyab3.1两端设计引物,扩增出的片段长为1029bp,引物序列为:
上游引物yab3.1-gfp-f:5'-aatgatcttcctaagccacctga-3'(seqidno.6),
下游引物yab3.1-gfp-r:5’-gtatagttcatccatgccatgtgta-3’(seqidno.7);
分别以卡那霉素抗性植株和未转化植株dna为模板,以yab3.1-gfp-f和yab3.1-gfp-r为引物,进行pcr检测,扩增产物进行琼脂糖凝胶电泳检测分析。
2)荧光定量rt-pcr检测:
提取抗卡那霉素植株叶片及未转化株叶片总rna,反转录成第一链cdna,建立荧光定量rt-pcr扩增体系,每个样品重复3次,根据数据分析得到各个样品的ct值,以未转化植株的表达为基准值,计算各转基因植株及野生型基因相对表达情况,特异引物扩增出的片段长为192bp,引物序列为:
上游引物cmyab3.1-rt-f:5′-cagtttcatttcggtccttcttta-3′(seqidno.8),
下游引物cmyab3.1-rt-r:5′-tctctgtctcttctctggtgctct-3′(seqidno.9);
以ef1α扩增的基因片段为内标,片段长度为151bp,引物序列:
上游引物ef1α-f:5′-ttttggtatctggtcctggag-3′(seqidno.10),
下游引物ef1α-r:5′-ccattcaagcgacagactca-3′(seqidno.11)。
具体的,上述的通过转cmyab3.1基因调控菊花株系的方法:
其在于步骤(3)中所述的采用农杆菌介导将cmyab3.1基因转入菊花的过程为:制备农杆菌eha105感受态,将步骤(2)中构建的cmyab3.1基因的植物表达载体pore-r4-35saa-cmyab3.1转入农杆菌eha105中,挑选阳性克隆,摇菌至od=0.5,离心后,弃上清,用ms(ph5.8)培养液将沉淀等体积悬浮,用于侵染;取组培瓶中菊花苗嫩叶叶盘作为转化受体,在预培养基中培养3d,然后浸入上述备好的转入了pore-r4-35saa-cmyab3.1的植物表达载体的农杆菌菌液中浸泡8~10min,用滤纸吸干叶盘表面的菌液后再将其接种到共培养基上,暗培养3d,然后转入筛选培养基上继代培养4代,等分化出的抗性芽长至2~3厘米时转入生根培养基上培养,初步获得抗性植株。所用的农杆菌菌株为eha105。
上述的通过转cmyab3.1基因调控菊花株型的方法,其在于菊花组织培养基以ms培养基为基础,ph5.8,100kpa、121℃灭菌20分钟;预培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;共培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;筛选培养基:ms+卡那霉素(kan)10mg/l+羧卞青霉素(carb)500mg/l+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.1mg/l;生根培养基:ms+卡那霉素(kan)8mg/l。
更详细的操作过程为:
取5μlpore-r4-35saa-cmyab3.1载体质粒,加入100μl感受态细胞,进行质粒转化农杆菌感受态后,挑取单克隆检测,选取阳性克隆摇菌,用于转化菊花。
以切花菊‘神马’叶片为外植体,取组培瓶中菊花苗嫩叶叶盘(0.5cmⅹ0.5cm)作为转化受体,预培养3d,然后浸入备好的农杆菌菌液中侵染8~10min,用滤纸吸干菌液后再接种到共培养基上黑暗中共培养3d,然后转入筛选培养基上继续培养,等分化出的抗性芽长至2~3厘米时转入生根培养基上培养,初步获得抗性植株。
上述的通过转cmyab3.1基因调控菊花株型的方法,其在于将转cmyab3.1基因初步获得的抗性植株进行pcr鉴定、荧光定量rt-pcr分子检测,筛选阳性植株,获得转基因菊花株系:(1)pcr检测
取生根筛选得到的卡那霉素抗性植株嫩叶及未转化株嫩叶,提取基因组dna,将gfp的一段序列和cmyab3.1的一段序列作为检测目标,在gfp和cmyab3.1两端设计引物,扩增出的片段长应为1029bp,引物序列为:
上游引物yab3.1-gfp-f:5'-aatgatcttcctaagccacctga-3'(seqidno.6),
下游引物yab3.1-gfp-r:5′-gtatagttcatccatgccatgtgta-3′(seqidno.7);
分别以卡那霉素抗性植株和未转化植株dna为模板,以yab3.1-gfp-f和yab3.1-gfp-r为引物,进行pcr检测,扩增产物进行琼脂糖凝胶电泳检测分析;阳性植株应得到长度为1029bp的产物,阴性植株则无扩增产物。
(2)荧光定量rt-pcr检测
提取抗卡那霉素植株叶片及未转化株叶片总rna,反转录成第一链cdna,建立荧光定量rt-pcr扩增体系,每个样品重复3次,根据数据分析得到各个样品的ct值,以未转化植株的表达量为基准值,计算各转基因植株及野生型基因相对表达情况;特异引物扩增出的片段长为192bp,引物序列为:
上游引物cmyab3.1-rt-f:5′-cagtttcatttcggtccttcttta-3′(seqidno.8),
下游引物cmyab3.1-rt-r:5′-tctctgtctcttctctggtgctct-3′(seqidno.9);以ef1α扩增的基因片段为内标,片段长度为151bp,引物序列:
上游引物ef1α-f:5′-ttttggtatctggtcctggag-3′(seqidno.10),
下游引物ef1α-r:5′-ccattcaagcgacagactca-3′(seqidno.11);
上述的通过转cmyab3.1基因调控菊花株型的方法,其在于对通过pcr、荧光定量rt-pcr分子检测所获得转基因植株后代进行表型观察:
选取野生型菊花(wt)和转基因菊花(ox-cmyab3.1)的扦插苗定植65天后作为表型观察的材料,每个株系20株苗。统计每个株系的高度和茎粗。
本发明的第三个目的是提供上述的基因cmyab3.1,或
上述的植物表达载体,或
上述的调控菊花株型的方法在调控菊花株型方面的基因工程应用。
进一步的,所述调控菊花株型是指将菊花株型改变为植株矮小,茎秆粗壮株型。
本发明所提供的通过转cmyab3.1基因调控菊花株型的方法具有如下优点:
本发明提供的方法选育了转基因菊花材料,采用转基因技术,将cmyab3.1基因整合到菊花基因组中,通过表型观察及分析获得植株矮小,茎秆粗壮的转基因菊花株系。因此利用本方法向菊花基因组中导入的cmyab3.1基因可以显著降低菊花高度,并使之粗壮。
附图说明
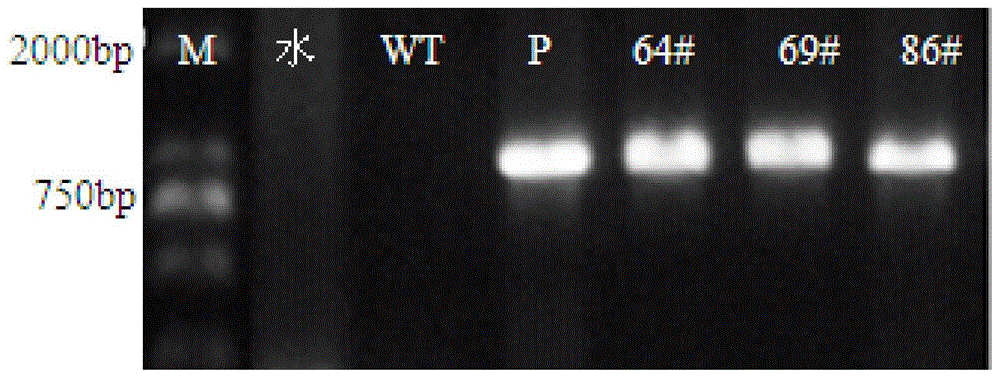
图1转cmyab3.1基因菊花特异引物检测电泳
m:dl2000marker;
wt:野生型植株;
p:pore-r4-35saa-cmyab3.1载体质粒
64#,69#,86#:分别为转入cmyab3.1过表达载体的菊花株系
图2转基因及野生型菊花植株中cmyab3.1基因的相对表达量
wt:野生型植株;
64#,69#,86#:分别为转入cmyab3.1过表达载体的菊花株系
图3野生型及转基因菊花植株表型观察
图a为野生型及转基因菊花侧面图
图b为野生型及转基因菊花俯视图
其中,wt:野生型植株;
ox-cm-yab3.1-64,ox-cm-yab3.1-69,ox-cm-yab3.1-86:分别为转入cmyab3.1过表
达载体的菊花株系
图4野生型及转基因菊花植株高度统计
wt:野生型植株;
64#,69#,86#:分别为转入cmyab3.1过表达载体的菊花株系
图5野生型及转基因菊花植株茎粗统计
wt:野生型植株;
64#,69#,86#:分别为转入cmyab3.1过表达载体的菊花株系
具体实施方式
下面对发明的具体实施例进行详细的说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,其具体实施方式如下:
实施例1.cmyab3.1的克隆
以切花大菊‘神马’为材料,取叶片0.15g,参照trizolrna提取试剂盒(takara)说明书的操作方法提取叶片的总rna,根据m-mlv反转录试剂盒(takara)进行反转录获得cdna,根据菊花转录组数据库中该基因的序列信息,运用primer5软件设计特异引物扩增cmyab3.1;
上游引物cmyab3.1-f:5′-atgtctaccatcacagccac-3′(seqidno.2),
下游引物cmyab3.1-r:5′-ctaataataaggagagacaccaac-3′(seqidno.3);
以叶片的cdna为模板,进行pcr反应,50μl反应体系:10×pcrbuffer5.0μl,cmyab3.1-f、cmyab3.1-r引物各1.0μl(20μmol·l-1),dntpmix4.0μl(2.5mmol·l-1),taqdnapolymerase0.2μl,cdna模板1μl,ddh2o37.8μl;反应程序:95℃预变性3min,然后94℃解链30sec,55℃退火30sec,72℃延伸1min,反应35个循环,72℃延伸10min;产物用凝胶回收试剂盒(axygen,usa)回收,用t4dna连接酶(takara)连接到pmd19-t载体(takara),转化dh5α感受态细胞,序列测定为seqidno.1。
实施例2.植物表达载体pore-r4-35saa-cmyab3.1的构建
根据cmyab3.1全长基因序列设计引物进行pcr反应,在目的基因cmyab3.1的上游和下游分别引入酶切位点xhoi和smai。以含seqidno.1序列的质粒作为模板,用高保真酶(primestartmhsdnapolymerase,takara)进行pcr反应,50μl反应体系:10×hspcrbuffer5.0μl,cmyab3.1-xhoi-f、cmyab3.1-smai-r引物各1.0μl(20μmol·l-1),dntpmix4.0μl(2.5mmol·l-1),primestartmhsdnapolymerase0.4μl,cdna模板1μl,ddh2o37.6μl;反应程序:95℃预变性5min,然后94℃解链30sec,55℃退火30sec,72℃延伸1min,反应35个循环,72℃延伸10min;pcr产物用凝胶回收试剂盒(axygen,usa)回收,再将回收的pcr产物和植物过表达载体pore-r4-35saa由xhoi和smai双酶切,对其酶切产物分别回收,再将其回收产物用连接酶连接后,转化dh5α感受态细胞,提取阳性质粒,植物表达载体pore-r4-35saa-cmyab3.1构建成功。
上游引物cmyab3.1-xhoi-f:5′-ccgctcgagatgtctaccatcaca-3′(seqidno.4),
下游引物cmyab3.1-smai-r:5′-tcccccgggataataaggagagac-3′(seqidno.5);
实施例3.农杆菌eha105介导叶盘法转化菊花
从yeb(50μg/ml利福平)平板上挑取eha105单菌落,接种于50ml含50μg/ml利福平的yeb液体培养基中,200rpm,28℃培养至od值0.5,而后冰浴菌液30min,离心收集菌体,悬浮于2ml预冷的100mmcacl2(20%甘油)溶液中,200μl/管分装,待用。取10μlpore-r4-35saa-cmyab3.1载体质粒,加入200μl感受态细胞,冰浴30min,液氮冷冻5min,37℃5min,加入800μlyeb液体培养基,28℃200rpm预培养4h,菌液涂板于yeb(50μg/ml利福平+50μg/ml卡那霉素)固体培养基上,28℃暗培养2天,挑取单克隆检测,选取阳性克隆摇菌,用于转化菊花。
采用的农杆菌指包含cmyab3.1基因的植物表达载体的eha105,用yeb液体培养基培养农杆菌eha105;将转基因植株后代命名为ox-yab3.1-n,以切花菊‘神马’叶片为外植体,采用根癌农杆菌介导法将‘神马’中克隆得到的cmyab3.1基因导入菊花。取组培瓶中菊花苗顶端叶盘(0.5cmⅹ0.5cm)作为转化受体,在预培养培养基中预培养3d,然后浸入备好的农杆菌菌液中浸泡10min,用滤纸吸干菌液后再接种到共培养培养基上黑暗中共培养3d,然后转入筛选培养基上,等分化出的抗性芽长至2~3厘米时转入生根培养基上培养,初步获得抗性植株。
菊花组织培养基以ms培养基为基础,ph5.8,100kpa、121℃灭菌20分钟;预培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;共培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;筛选培养基:ms+卡那霉素(kan)10mg/l+羧卞青霉素(carb)500mg/l+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.1mg/l;生根培养基:ms+卡那霉素(kan)8mg/l。
实施例4.对转cmyab3.1基因的抗性植株进行分子检测(pcr、荧光定量rt-pcr),获得阳性转基因菊花株系
1)pcr检测
取生根筛选得到的卡那霉素抗性植株嫩叶及未转化株嫩叶,提取基因组dna,将gfp的一段序列和cmyab3.1的一段序列作为检测目标,在gfp和cmyab3.1两端设计引物,扩增出的片段长为1029bp,引物序列为:
上游引物yab3.1-gfp-f:5'-aatgatcttcctaagccacctga-3'(seqidno.6),
下游引物yab3.1-gfp-r:5’-gtatagttcatccatgccatgtgta-3’(seqidno.7);
分别以卡那霉素抗性植株和未转化植株dna为模板,以yab3.1-gfp-f和yab3.1-gfp-r为引物,进行pcr检测。
扩增体系为:1μldna模板,2.5μl10×pcrbuffer,2μldntp,1.5μl2.5mmol·l-1mgcl2,5u·μl-1taq酶0.2μl,yab3.1-gfp-f和yab3.1-gfp-r各1μl,去离子水补足体积至25μl。
扩增条件为:94℃预变性5min,94℃变性1min,58℃退火45s,72℃延伸70s,30个循环;72℃延伸10min。扩增产物进行琼脂糖凝胶电泳检测分析(如图1)。
图中可以看到,转cmyab3.1的植株均可扩增出与以pore-r4-35saa-cmyab3.1质粒为模板的阳性对照相同的特异条带,而野生型植株没有扩增出条带。
2)荧光定量rt-pcr检测
提取抗卡那霉素植株叶片及未转化株叶片总rna,反转录成第一链cdna,用荧光定量rt-pcr对cmyab3.1基因的表达量进行检测。按照荧光定量试剂盒(
上游引物cmyab3.1-rt-f:5′-cagtttcatttcggtccttcttta-3′(seqidno.8),
下游引物cmyab3.1-rt-r:5′-tctctgtctcttctctggtgctct-3′(seqidno.9);以ef1α扩增的基因片段为内标,片段长度为151bp,引物序列:
上游引物ef1α-f:5′-ttttggtatctggtcctggag-3′(seqidno.10),
下游引物ef1α-r:5′-ccattcaagcgacagactca-3′(seqidno.11);
上述的通过转cmyab3.1基因调控菊花株型的方法,其在于对通过pcr、荧光定量rt-pcr分子检测所获得转基因植株后代进行表型观察:
根据荧光定量rt-pcr检测结果(图2),与野生型相比,转基因株系64#、69#、86#株系表达量明显升高。证实cmyab3.1基因已经导入切花菊基因组dna中并表达。
实施例5.转基因植株后代的表型观察
选取野生型菊花(wt)和转基因菊花(ox-cmyab3.1)的扦插苗定植65天后作为表型观察的材料,观察到转基因株系ox-cmyab3.1-64#、ox-cmyab3.1-69#、ox-cmyab3.1-86#明显矮于野生型株系(图3),并对野生型及转基因株系的株高和茎粗进行测量与统计。
野生型株系的平均高度为77.53cm,转基因株系64#、69#、86#的平均高度分别为25.07cm、20.53cm、23.27cm,均只有野生型株系高度的0.32倍以下(图4)。野生型株系的平均茎粗为6.01cm,转基因株系64#、69#、86#的平均茎粗分别为8.07cm、7.74cm、7.75cm,为野生型株系茎粗的1.28倍以上(图5)。利用spss软件进行独立样本t检验发现野生型株系与转基因株系的高度有极显著性差异。转基因植株表型观察实验表明转入cmyab3.1基因对菊花植株高度具有明显的抑制作用,并促进茎粗的增加,从而改变菊花的株型,使之矮壮。
序列表
<110>南京农业大学
<120>一种创建矮壮株型菊花的方法
<160>11
<170>siposequencelisting1.0
<210>1
<211>684
<212>dna
<213>菊花‘神马’(chrysanthemummorifolium'jinba')
<400>1
atgtctaccatcacagccaccaccttaccaccatcaccaccacaagcacccctaaacaac60
accgctgatcagctttgctatgttcattgcacccgctgcgacaccgtcctagcggttagt120
gttccttgtacaagcttgttcaagaccgtgaccgtacgatgtggtcattgcaccaacctc180
ttgccagttaacatgcgtgggttgatgctaccaactccggcggttaatcagtttcatttc240
ggtccttctttattctctccttccacgcatggttttcaggatgattggataacaaatggg300
aactcgaacttttcgatgaatcagaattacccaagagatgtttctataacatcttcaaga360
ggatttaatgatcttcctaagccacctgagatcaacagagcaccagagaagagacagaga420
gtcccatcggcctacaaccgtttcatcaaggaagaaattcagaggatcaaagccagaaat480
cccgatatcagtcatagagaagcgtttagtgctgctgccaaaaactgggctcactttcca540
cacattcactttggtctcatggctgatcatcagactgctgctgctgccaacaggaccagc600
atgcgccaacaggagattgaaggtggcgtcatgaaagaaaacttatttgctgctgcaaat660
gttggtgtctctccttattattag684
<210>2
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgtctaccatcacagccac20
<210>3
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>3
ctaataataaggagagacaccaac24
<210>4
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>4
ccgctcgagatgtctaccatcaca24
<210>5
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>5
tcccccgggataataaggagagac24
<210>6
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>6
aatgatcttcctaagccacctga23
<210>7
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>7
gtatagttcatccatgccatgtgta25
<210>8
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>8
cagtttcatttcggtccttcttta24
<210>9
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>9
tctctgtctcttctctggtgctct24
<210>10
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>10
ttttggtatctggtcctggag21
<210>11
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>11
ccattcaagcgacagactca20
- 还没有人留言评论。精彩留言会获得点赞!