一种提高5-氯-1-茚酮收率的合成方法与流程
本发明属于有机合成的技术领域,具体涉及一种提高5-氯-1-茚酮收率的合成方法。
背景技术:
5-氯-1-茚酮是一种重要的有机化工中间体,广泛应用与农药、医药等化工品的合成中。以5-氯-1-茚酮为原料生产的茚虫威(indoxacard)等新型杀虫剂具有高效低残留等特性,是国家产业政策鼓励发展的项目,具有十分广阔的应用前景。据文献报道,该化合物主要合成路线有以下三种:
路线一:间氯苯甲醛法
路线一是以间氯苯甲醛为主原料,历经缩合、催化加氢、傅克烷基化等反应,合成步骤相对繁琐。并且在催化加氢时要用到钯炭,生产成本较高;关环过程选择性差,易形成7-氯-1-茚酮,其性质与产品相似,造成分离困难,因此该路线难以进行工业化生产。
路线二:3-氯苄基氯法
路线二是以3-氯苄基氯为主原料,经取代、水解、脱羧、氯化和傅克酰基化等5步反应合成5-氯-1-茚酮,合成步骤繁琐、复杂,从成本角度考虑不易于工业化生产。
路线三:3,4’-二氯苯丙酮法
其中3,4’-二氯苯丙酮合成,由以下两种方法:
方法一:以三氯丙酰氯为主原料,在三氯化铝催化作用下,与氯苯发生傅克酰基化反应,合成得到。
方法二:以对氯苯甲酰氯为主原料,在三氯化铝催化作用下,与乙烯发生傅克酰基化反应,合成得到。
路线三是以3,4’-二氯苯丙酮为主原料,经傅克烷基化反应,一步合成得到5-氯-1-茚酮。该方法合成步骤简单,但是所用催化剂为三氯化铝、三氟甲磺酸、浓硫酸等,反应条件苛刻,选择性差,路线三的收率仅为60~70%。
技术实现要素:
针对现有技术中目前傅克烷基化反应选择性差、副反应多、收率低等技术问题,本发明提供一种提高5-氯-1-茚酮收率的合成方法,以解决上述问题。本发明采用了路线三,以3,4’-二氯苯丙酮为原料,加入非质子酸和相转移催化剂,加热反应2~5h,反应结束后降温,倒入冰水中;过滤后固体经过有机溶剂纯化得到5-氯-1-茚酮。通过在反应中加入相转移催化剂,显著改善了反应体系的粘度、有效提高了傅克烷基化反应选择性、降低了副反应,提高了5-氯-1-茚酮合成收率。
本发明的技术方案为:一种提高5-氯-1-茚酮收率的合成方法,具体制备步骤如下:
(1)称取3,4’-二氯苯丙酮、非质子酸催化剂置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至80~100℃,体系呈现熔融状态;熔融状态可根据搅拌阻力变化及观测判断;
(2)待体系呈现熔融状态时,称取相转移催化剂,加入到反应瓶中,继续升温,0.5~1h升温至150~180℃;到达目标温度后保温反应2~5h,用hplc监测反应,当反应液中3,4’-二氯苯丙酮残留≤0.1%时,停止反应;
(3)将反应液降温至70~80℃,转入0~5℃水中,搅拌0.5~1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品;
(5)将步骤(4)所得粗品置于反应瓶中,加入有机溶剂及活性炭,搅拌加热至回流脱色0.5~1h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1~2h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮。
优选的,所述步骤(1)中非质子酸催化剂为alcl3、zncl2、incl3、ticl4、bf3、fe(cf3so3)3、fe(cf3so3)2、γ-氧化铝中的一种。
优选的,所述步骤(1)中非质子酸用量为以3,4’-二氯苯丙酮计摩尔比2.5~7:1,优选3:1。
优选的,所述步骤(2)中相转移催化剂为四丁基溴化铵、四丁基氯化铵、三辛基甲基氯化铵、苄基三乙基氯化铵、十二烷基三甲基氯化铵、十八冠六、十五冠五、聚乙二醇400、聚乙二醇600、聚乙二醇800。
优选的,所述步骤(2)中相转移催化剂为四丁基溴化铵、四丁基氯化铵或苄基三乙基氯化铵中的一种。
优选的,所述步骤(2)中相转移催化剂的用量为以3,4’-二氯苯丙酮重量计0.5~10%。
优选的,所述步骤(3)中水的用量为以3,4’-二氯苯丙酮计30g/g。
优选的,所述步骤(5)中有机溶剂为甲醇、乙醇、丙酮、石油醚、四氯化碳、乙酸甲酯、乙酸乙酯。
优选的,所述步骤(5)中有机溶剂用量为以步骤(4)所得粗品重量计2~5g/g。
优选的,所述步骤(5)中活性炭的用量为以步骤(4)所得粗品重量计5~10%。
本发明的有益效果为:
(1)本发明通过在傅克反应中加入相转移催化剂,明显的提高了傅克反应的选择性,降低了副反应的发生,提高了反应收率,经多批次实验结果可知,反应收率可提高至85%以上,产品纯度均在99%以上。
(2)本发明通过在傅克反应中加入相转移催化剂,明显的改善了反应体系的粘度,使反应后处理变得更加简单,方便;本发明在后处理的方式上均为本领域最基本的操作,因此有着极高的工业化生产前景。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
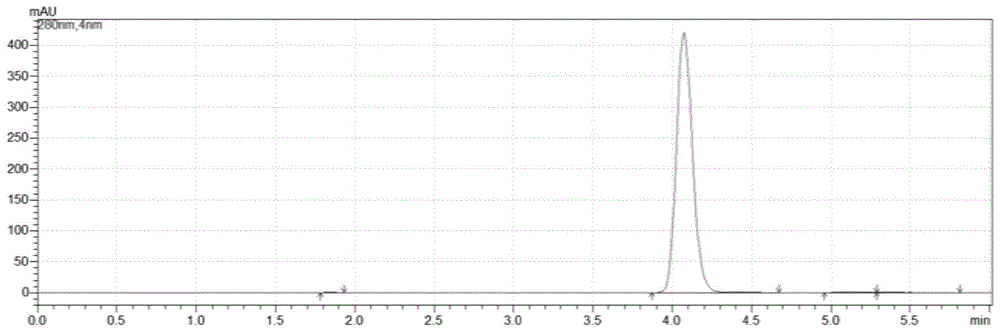
图1是本发明实施例1制备的5-氯-1-茚酮的hplc图谱;
图2是5-氯-1-茚酮标准品的hplc图谱;
图3是本发明实施例2制备的5-氯-1-茚酮的hplc图谱;
图4是本发明实施例3制备的5-氯-1-茚酮的hplc图谱;
图5是本发明实施例4制备的5-氯-1-茚酮的hplc图谱;
图6是本发明实施例5制备的5-氯-1-茚酮的hplc图谱;
图7是本发明实施例6制备的5-氯-1-茚酮的hplc图谱;
图8是本发明实施例7制备的5-氯-1-茚酮的hplc图谱;
图9是本发明实施例8制备的5-氯-1-茚酮的hplc图谱。
具体实施方式
为了使本技术领域的人员更好地理解本发明中的技术方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
实施例1
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g四丁基溴化铵,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应2h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品337.2g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮161.1g,收率:92.4%,纯度(面积归一法):99.268%。实施例1制备的5-氯-1-茚酮hplc检测图谱详见图1,检测结果如下表1:
表1-实施例1产品检测结果
将外购5-氯-1-茚酮标准品在相同的检测方法下进行检测,hplc检测图谱详见图2,检测结果如下表2:
表2-5-氯-1-茚酮标准品检测结果
由表1和表2的检测结果可知,本发明制备的产品与5-氯-1-茚酮标准品在hplc中保留时间一致,因此可证明本发明所制备的产物为5-氯-1-茚酮。
实施例2
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取1.06g四丁基溴化铵,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应4.5h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品311.8g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮154.0g,收率:88.1%,纯度(面积归一法):99.048%。实施例制备的5-氯-1-茚酮hplc检测图谱详见图3,检测结果如下表3:
表3-实施例2产品检测结果
实施例3
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g四丁基氯化铵,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应4h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品317.6g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1.5h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮154.6g,收率:89.2%,纯度(面积归一法):99.891%。实施例3制备的5-氯-1-茚酮hplc检测图谱详见图4,检测结果如下表4:
表4-实施例3产品检测结果
实施例4
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g苄基三乙基氯化铵,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应4h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品321.8g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮156.6g,收率:89.8%,纯度(面积归一法):99.314%。实施例4制备的5-氯-1-茚酮hplc检测图谱详见图5,检测结果如下表5:
表5-实施例4产品检测结果
实施例5
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g十二烷基三甲基氯化铵,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应4h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品309.5g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮153.6g,收率:88.2%,纯度(面积归一法):99.440%。实施例5制备的5-氯-1-茚酮hplc检测图谱详见图6,检测结果如下表6:
表6-实施例5产品检测结果
实施例6
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g三辛基甲基氯化铵,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应3h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品307.6g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮151.5g,收率:87.2%,纯度(面积归一法):99.615%。实施例6制备的5-氯-1-茚酮hplc检测图谱详见图7,检测结果如下表7:
表7-实施例6产品检测结果
实施例7
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g十八冠六,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应4h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品308.2g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮152.7g,收率:87.8%,纯度(面积归一法):99.532%。实施例7制备的5-氯-1-茚酮hplc检测图谱详见图8,检测结果如下表8:
表8-实施例7产品检测结果
实施例8
一种5-氯-1-茚酮的合成方法,具体步骤如下:
(1)称取211.0g3,4’-二氯苯丙酮、400.5g三氯化铝置于反应瓶中,连接尾气吸收装置,开启搅拌,缓慢升温至100℃,体系呈现熔融状态;
(2)待体系呈现熔融状态时,称取21.1g聚乙二醇400,加入到反应瓶中,继续升温,1h升温至160℃;到达目标温度后保温反应4h,用hplc监测反应,此时无3,4’-二氯苯丙酮剩余,停止反应;
(3)将反应液降温至80℃,转入0~5℃水中,搅拌1h;
(4)抽滤,滤饼用水淋洗至滤液ph=6~7,抽滤至无水珠下落,得到粗品305.6g;
(5)将步骤(4)所得粗品置于反应瓶中,加入700g甲醇和25g活性炭,搅拌加热至回流脱色0.5h;
(6)脱色完毕,热过滤,滤液加热至复溶后缓慢降温至0~5℃,搅拌1h,抽滤至无液滴下落得湿品,烘干得5-氯-1-茚酮148.2g,收率:85.1%,纯度(面积归一法):99.420%。实施例8制备的5-氯-1-茚酮hplc检测图谱详见图9,检测结果如下表9:
表9-实施例8产品检测结果
由实施例1~8的收率及hplc检测结果可知,本发明的合成方法,反应收率有着明显的提升,并且制备产品的纯度均在99%以上。并且本发明在后处理的方式上均为本领域最基本的操作,有着极高的工业化生产前景。
尽管通过参考附图并结合优选实施例的方式对本发明进行了详细描述,但本发明并不限于此。在不脱离本发明的精神和实质的前提下,本领域普通技术人员可以对本发明的实施例进行各种等效的修改或替换,而这些修改或替换都应在本发明的涵盖范围内/任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求所述的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!