肽类化合物及其制备方法与应用与流程
本发明涉及属于药物化学技术领域,尤其是涉及一种肽类化合物及其制备方法与应用。
背景技术:
海绵是最古老最简单的多细胞动物,有5亿多年的演化史,依靠过滤海水取食,缺乏真正的组织,不能移动并缺乏物理防御。在海洋严酷的竞争环境中,海绵通过产生活性次级代谢产物来防御外来生物攻击。海绵是海洋新化合物最多产和最重要的来源之一,自20世纪60年代开始研究海洋天然产物以来,每年从海绵中发现结构新颖的活性物质约占海洋天然产物的三分之一。目前国内外共有49个来源于海洋的活性物质被批准上市或进入临床,其中有14个源自海绵(上市3个),因此,海绵已成为海洋药物的重要来源,一直是海洋天然产物化学家的研究热点。
海绵是海洋活性肽的重要来源,主要包括线性肽,缩酚酸肽和环肽。其中环肽类化合物是一类以酰胺键形成的环状化合物,主要是由氨基酸、糖肽、脂肽和核苷肽经过一系列修饰后组合而成的复杂分子。环肽类化合物作为药物分子,具有抗癌、抗病毒、抗菌、免疫抑制等广泛的生物活性。因此,针对环肽类药物的研究正在受到人们越来越多的关注。目前共有40多种环肽类药物用于临床,近十年,平均每年约有一种环肽类药物批准上市,占同时期上市新药总数的3%。环肽类化合物之所以在药物研发领域大展身手,与其几大特性相关。首先,由于环状结构导致的构象变化限制,环肽类化合物一般具有较大的表面积,从而使其与靶点蛋白间具有高度亲和力及识别特异性。大环结构构象灵活性的限制也降低了药物与靶点结合的熵值,提高了结合的稳定性。其次,氨基酸组成特点决定了环肽类化合物往往具有极低的、甚至没有细胞毒性。第三,环肽类化合物很容易通过自动化的化学合成流程实现生产,并且便于进行各项修饰、处理及监控,这些特点都非常有利于药物开发过程。
尽管环肽类化合物具有广阔的市场应用前景,但海绵来源的环肽类化合物在生物体内大多含量低微,其定向获取极富挑战。若单靠活性指导下的追踪分离往往由于对特异性成分的灵敏度不够,易产生错误导向,且步骤复杂,费时费力,通量低,容易导致微量活性成分的遗漏,极大限制了海绵中含量低微、结构新颖、活性显著的环肽类天然产物的发现进程。近年来,由于质谱技术的快速发展,极大推动了海洋天然产物的发现速度。特别是复合型液相串联三重四极杆质谱(q-q-q),通过母离子扫描、子离子扫描、中性丢失扫描和多离子反应监测等功能,大大提高了从复杂天然提取物中发现微量活性成分的灵敏度及选择性。其中,中性丢失扫描是指q1和q3同时进行全扫描,但是二者始终保持一定固定的质量差,只有在碰撞池q2中丢失的中性部分满足这个固定质量差的离子才能被检测到。基于环肽类化合物丢失固定中性碎片的特点结合质谱中性丢失扫描功能,建立环肽类特征结构的质谱快速定位方法,快速筛选出复杂提取物中含有该骨架片段的新颖环肽类化合物,这将为海洋来源复杂化合物的相关研究提供新的研究思路,加快海洋药物开发的速度。
技术实现要素:
本发明的第一目的在于公开一种肽类化合物,其为具有肿瘤细胞抑制活性的线性肽或环肽,其含有以下犬尿氨酸和精氨酸的缩合片段:
较佳的,其由3~20优选4~12个氨基酸组成。
进一步的,其为以下化合物1~6中的一种:
本发明的第二目的在于提供两种制备所述的肽类化合物的方法。
其中一种制备方法是从海绵中提取并分离制得,本发明的提取分离方法包括提取、萃取、分离富集、色谱纯化等步骤。
具体的一些实施例中,包括以下步骤:
(a)提取:将海绵粉碎后,用有机溶剂渗漉提取,合并提取液后减压浓缩得到提取液;所述有机溶剂优选乙醇、甲醇、丙醇等醇类溶剂或者两种及以上醇类溶剂的结合;
(b)萃取分离:提取液用乙酸乙酯萃取,乙酸乙酯层浓缩得到的浸膏混悬于80~90%优选85~90%的甲醇-水中,用石油醚萃取;将80~90%优选85~90%的甲醇-水层稀释至50~60%优选55%~60%的甲醇-水,用二氯甲烷萃取,合并萃取液并减压浓缩得到二氯甲烷萃取部位;
(c)分离富集:采用质谱追踪定位分析,通过凝胶柱层析、反相中压柱色谱和全制备高效液相色谱中的一种或多种方式分离富集含有大分子量化合物的馏分;
(d)筛选目标化合物:采用中性丢失扫描质谱法筛选含犬尿氨酸的目标化合物,首先检测馏分中中性丢失190da的母离子,然后对其进行子离子扫描验证,二级碎片离子中能发现190da的质量差的即为目标化合物母离子,进而获得其碎片离子信息和色谱保留行为;
(e)质谱导向分离:在获得其碎片离子信息和色谱保留行为基础上,采用质谱导向的半制备高液相色谱法对馏分进行分离,得到目标化合物。
更具体的,对于化合物1,可通过以下步骤从海绵中提取:
(a)提取:将棕色扁海绵粉碎后,用乙醇渗漉提取,合并提取液后减压浓缩得到提取液;
(b)萃取分离:提取液用等体积乙酸乙酯萃取3~5次优选3次,合并乙酸乙酯层后浓缩得到的浸膏混悬于80~90%优选85~90%的甲醇-水中,用等体积的石油醚萃取3~5次;将80~90%优选85~90%的甲醇-水层稀释至50~60%的甲醇-水,用等体积的二氯甲烷萃取3~5次优选3次,合并萃取液并减压浓缩得到二氯甲烷萃取部位;
(c)分离富集:将上述二氯甲烷萃取部位通过sephadexlh-20凝胶柱层析,用50%的ch2cl2-meoh洗脱剂洗脱,并采用质谱定位追踪的方式,将大分子量的化合物进行富集得;采用ods中压柱色谱分离,用meoh/h2o梯度(10%-100%,450min)洗脱,采用质谱追踪定位分析,获得含有大分子量的环肽类化合物的精细馏分;
(d)筛选目标化合物:采用中性丢失扫描质谱法筛选含犬尿氨酸的目标化合物,首先检测馏分中中性丢失190da的母离子m/z884.48[m+h]+,对其进行子离子扫描验证,二级碎片离子中能发现190da的质量差,进而获得其碎片离子信息和色谱保留行为;
(e)质谱导向分离:在获得其碎片离子信息和色谱保留行为基础上,采用质谱导向的半制备高液相色谱法对馏分进行分离,得到目标化合物。
另一种制备方法是全合成方法,包括以下步骤:
(a)按照肽链上的氨基酸序列,将第一种氨基酸装载到2-ctc树脂中:将2-ctc树脂溶胀后,加入fmoc(9-芴基甲氧基羰基保护基)保护的氨基酸和diea(n,n-二异丙基乙胺),反应完全后洗涤去除未反应的原料;
(b)fmoc脱保护:在室温下,使用含15~30%优选20%哌啶的dmf溶液进行fmoc脱保护,然后将树脂用dmf洗涤;
(c)肽偶联:按照肽链上的氨基酸序列,依次通过以下步骤偶联其余氨基酸:将fmoc保护的氨基酸、hatu(多肽缩合试剂,系统命名为2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)和diea在dmf中室温下与树脂轻轻涡旋搅拌,然后将树脂用dmf洗涤;其余氨基酸中的精氨酸和色氨酸的侧链分别由pbf基团和boc基团保护;
(d)fmoc脱保护:在室温下,使用含15~30%优选20%哌啶的dmf溶液进行fmoc脱保护,然后将树脂用dmf洗涤;
(e)裂解:过滤树脂,并用hfip裂解溶液裂解,裂解完成后纯化得到侧链保护的线性肽;
(g)侧链脱保护:向含保护基的线性肽中加入裂解混合物进行脱保护,反应完全后纯化得到前体肽;按体积百分比计,所述裂解混合物含有90~95%的tfa(三氟乙酸)、2.5~5%的tis(三异丙基硅烷)和2.5~5%的h2o,优选93~95%的tfa、2.5~3.5%的tis和2.5~3.5%的h2o。
(h)臭氧氧化:将前体肽溶于meoh中,冷却至-78℃,使用臭氧发生器将o3鼓入反应4~8分钟;在-78℃下将二甲基硫醚添加到反应中,使反应混合物在0.8~1.2小时内升温至室温;将反应混合物在真空下浓缩,然后溶于的tfa/h2o的混合物中,并在室温下搅拌0.8~1.2小时得到目标化合物;
或者,在步骤(g)之前,其还包括步骤:
(f)大环化:线性肽在dcm(二氯甲烷)中的溶液加入edci((1-乙基-3(3-二甲基丙胺)碳二亚胺),hoat(1-羟基-7-氮杂苯并三氮唑)和diea,室温搅拌反应进行关环,然后真空除去溶剂。
以化合物1为例,可采用以下合成路线制备:
具体包括以下步骤:
(a)将第一种氨基酸装载到2-ctc树脂中:将2-ctc树脂在装有无水dcm的一次性容器中溶胀15~30分钟;加入fmoc保护的脯氨酸(1.5~2.5个当量)和diea(3~5个当量)的dcm溶液,并将反应容器在室温下在涡旋中摇动0.8~1.5小时;向反应混合物中加入meoh,并将树脂旋转10~20分钟;过滤树脂并用dcm(3ml×5次,1分钟/次),dcm/meoh(v/v=1∶1)(3ml×5次,1分钟/次)和meoh(3ml×2次,1分钟/次)洗涤;
(b)fmoc脱保护:在室温下,使用含15~30%哌啶的dmf溶液进行fmoc脱保护15~30分钟,然后将树脂用dmf洗涤(3ml×2次,1分钟/次);
(c)肽偶联:按照肽链上的氨基酸序列,依次通过以下步骤偶联其余氨基酸:将fmoc保护的氨基酸(2~4个当量)、hatu(2~4个当量)和diea(5~7个当量)在dmf中室温下与树脂轻轻涡旋搅拌0.8~1.2小时,然后将树脂用dmf洗涤(3ml×5次,1分钟/次);其余氨基酸中的精氨酸和色氨酸的侧链分别由pbf基团和boc基团保护;
(d)fmoc脱保护:在室温下,使用含15~30%哌啶的dmf溶液进行fmoc脱保护15~30分钟,然后将树脂用dmf洗涤(3ml×2次,1分钟/次);
(e)裂解:过滤树脂,并用3ml20%(v/v)hfip裂解溶液在无水dcm中处理0.8~1.5小时,然后重复此步骤处理30分钟;过滤后,合并所得裂解溶液,真空浓缩,通过hplc纯化,得到侧链保护的线性肽1a;
(f)大环化:将线性肽1a(1当量)在dcm中的溶液加入edci(3当量),hoat(3当量)和diea(3当量),将该溶液在室温搅拌16h,然后真空除去溶剂;
(g)侧链脱保护:向含保护基的环肽中加入裂解混合物(tfa:tis:h2o=95/2.5/2.5,v/v/v),搅拌3h,通过lc-ms监测反应;将该溶液真空浓缩,然后通过hplc纯化,得到前体肽1b;
(h)臭氧氧化:将前体肽1b溶于meoh中,冷却至-78℃,使用臭氧发生器将o3鼓入反应5分钟;在-78℃下将二甲基硫醚添加到反应中,使反应混合物在1小时内升温至室温;将反应混合物在真空下浓缩,然后溶于的tfa/h2o(1/1,v/v)的混合物中,并在室温下搅拌1h得到目标化合物1。
本发明的第三目的在于公开所述的肽类化合物在制备抗肿瘤药物中的应用。
本发明的肽类化合物对mcf-7、hela、nci-h460、pc9这四株肿瘤细胞显示出很强的抑制活性,其中化合物1对上述四株肿瘤细胞的ic50值分别为3.4、6.2、7.1和12μm,同阳性药顺铂活性相当,且对正常细胞h9c2毒性很低ic50>100μm,作用机制研究发现化合物1为rxrα的潜在拮抗剂通过pi3k/akt信号通路抑制癌细胞的增殖并促进细胞凋亡。构效关系分析,kyn和胍基为本发明化合物的药效团,并通过分子对接实验验证了其作用靶点和药效基团。因此本发明化合物可用于制备抗肿瘤药物。
本发明的积极进步效果在于:
本发明提供一种从复杂提取物中选择性识别含犬尿氨酸的肽类化合物的质谱快速定位方法。从肽化合物的质谱裂解机制中发现,环肽经碰撞诱导解离后形成的二级碎片离子往往是丢失某种中性分子片段导致的,这些中性分子大多是组成该环肽的基本单元——各氨基酸残基。根据这一特点,分析二级ms发现,可以通过碎片离子间含190da的质量差的母离子快速寻找含犬尿氨酸的环肽。运用中性丢失质谱法扫描从中国西沙群岛附近海域采集的棕色扁海绵phakelliafusca的粗馏分,从中发现母离子为m/z884.48[m+h]+的峰,在获得其碎片离子信息和色谱保留行为基础上,结合质谱引导制备液相将其分离,得到一种新的环肽类化合物。
本发明运用固相合成,液相环化及最后阶段的臭氧化策略实现了对本发明化合物的全合成,为其后续研究提供了量的保证。
本发明化合物对mcf-7、hela、nci-h460、pc9这四株肿瘤细胞显示出很强的抑制活性,其中化合物1对四株肿瘤细胞的ic50值分别为3.4、6.2、7.1和12μm,同阳性药顺铂活性相当,且对正常细胞h9c2毒性很低ic50>100μm,作用机制研究发现化合物1为rxrα的潜在拮抗剂通过pi3k/akt信号通路抑制癌细胞的增殖并促进细胞凋亡。构效关系分析,kyn和胍基为本发明化合物的药效团,并通过分子对接实验验证了其作用靶点和药效基团。因此本发明化合物可用于制备抗肿瘤药物。
本发明化合物制备方法简单,对肿瘤细胞抑制活性显著。本发明为研究和开发新抗肿瘤药物提供了新的先导化合物,为微量环肽类细胞毒活性成分快速识别和定向追踪提供了新方法,为环肽类化合物的合成提供了新思路。为开发利用我国海洋药用资源提供了科学依据。
附图说明
图1是天然化合物1的发现流程图;
图2是天然化合物1的ms/ms碎片检测图;
图3是天然化合物1通过lc-ms的高级marfey’s分析图;
图4是合成与天然的化合物1的1hnmr(dmso-d6,600mhz)谱对比图;
图5是合成与天然的化合物1的13cnmr(dmso-d6,150mhz)谱对比图;
图6是合成与天然的化合物1的ms/ms碎片对比图;
图7是不同浓度化合物1诱导hela细胞凋亡图;
图8是不同浓度化合物1诱导hela细胞周期阻滞图;
图9是化合物1对rxrα的影响及蛋白印迹分析图;
图10是化合物1与rxrα分子对接结果图。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
实施例1从海绵中发现并提取天然化合物1(c45h61n11o8)
从肽化合物的质谱裂解机制中发现,环肽经碰撞诱导解离后形成的二级碎片离子往往是丢失某种中性分子片段导致的,这些中性分子大多是组成该环肽的基本单元——各氨基酸残基。根据这一特点,分析二级ms发现,可以通过碎片离子间含190da的质量差的母离子快速寻找含犬尿氨酸的环肽。运用中性丢失质谱法扫描从中国西沙群岛附近海域采集的棕色扁海绵phakelliafusca(属于寻常纲(demospongiae)软海绵目(halichondrid)轴海绵科(axinellidae)棕色扁海绵属(phakellia))的粗馏分,从中发现母离子为m/z884.48[m+h]+的峰,如图1所示,在获得其碎片离子信息和色谱保留行为基础上,结合质谱引导制备液相将其分离,得到一种新的环肽类化合物,即化合物1。
中性丢失扫描质谱法在本发明化合物筛选中的应用具体如下:
1)共有中性片段的确定:
结合含犬尿氨酸环肽的二级质谱及犬尿氨酸残基的结构,确定中性片段的质量为190da,将“中性丢失扫描”方式的质量差设为190da。
2)“中性丢失扫描”的优化:
输入电压、碰撞能量等的优化,获得尽可能多的高响应度碎片离子信息,并确保能够找到母离子,一般母离子为碎片离子响应度的1/3为宜;设定筛选标准,设定质量差为190da作为标准,保证具有该特定质量差的离子通过。
3)样品前处理条件的优化:
通过sephadexlh-20凝胶柱层析,用50%的ch2cl2-meoh洗脱剂洗脱,并采用质谱定位追踪的方式,将大分子量的化合物主要富集;经历反相中压柱色谱及质谱导向的全制备高液相色谱,有效的去除杂质成分,富集环肽类成分。
4)液相条件的优化:
为使获得的特异成分实现基线分离,减少离子间干扰,便于多级离子结构分析和特异性成分的定向获取,确定最佳的线性梯度条件,尽量避免共流出现象的发生。
5)二级质谱验证:
在环肽类化合物粗馏分中检测到中性丢失190da母离子m/z884.48[m+h]+,对其进行子离子扫描验证,二级碎片离子中能发现190da的质量差。
6)质谱引导制备分离:
在获得其碎片离子信息和色谱保留行为基础上,结合质谱引导制备液相将m/z884.48[m+h]+分离。
从海绵中提取天然化合物1的具体步骤如下:
棕色扁海绵(干重1.0kg)粉碎后,用95%的乙醇渗漉提取,合并提取液后减压浓缩,回收乙醇,浓缩得到提取液。
提取液用等体积乙酸乙酯萃取三次,合并乙酸乙酯层后浓缩得到乙酸乙酯层浸膏(34.5g)。将乙酸乙酯层浸膏混悬于90%的甲醇水中,用等体积石油醚萃取三次,合并萃取液并减压浓缩得到石油醚层(22.5g)。将90%的甲醇水层稀释至60%的甲醇水,用等体积的二氯甲烷萃取三次,合并萃取液并减压浓缩得到二氯甲烷层(3.5g)。
将上述二氯甲烷萃取部位3.5g,通过sephadexlh-20凝胶柱层析,用50%的ch2cl2-meoh洗脱剂洗脱,并采用质谱定位追踪的方式,将大分子量的化合物进行富集得2.1g。采用ods中压柱色谱分离,用meoh/h2o梯度(10%-100%,450min)洗脱,采用质谱追踪定位分析,获得含有大分子量的环肽类化合物的精细馏分;最终采用质谱导向的半制备高液相色谱法对馏分进行分离(50-60%甲醇-水,流速5.0ml/min,在正离子模式下检测分子离子884.5),得到本发明化合物1。
通过上述步骤制得的天然化合物1的理化性质和核磁共振数据如下:
化合物1:淡黄色无定型粉末;[α]25d-71.0(c0.10,meoh);uv(meoh)λmax(logε)204(4.34)nm;ir(atr)νmax3326,2959,2930,2874,1616,1525,1446,1348,1245,1203,1162,1099,1042,748,701,544,484cm-1;hresimsm/z884.4788[m+h]+(calcdforc45h61n11o8,884.4783)。
核磁共振谱数据见表1。
表1:化合物1的核磁共振谱数据(dmso-d6)
asequentialnoes.boverlappingsignals.
通过tocsy谱,可以确定6个氨基酸的自旋偶合系统,包括一个苯丙氨酸、一个缬氨酸、一个精氨酸和三个脯氨酸,并通过hmbc谱验证。其余的信号证明了一个非常见氨基酸犬尿氨酸的存在。13cnmr谱中存在1个酮羰基信号δc199.0(kyn-γ-co)和1个sp2杂化的非质子碳δc151.4(kyn-c-6);1hnmr谱中δh7.64(2h,d,j=9.0hz,kyn-nh,kyn-h-2)和6.79(1h,d,j=8.4hz,kyn-h-5)为双重峰,δh6.57(1h,d,j=7.7hz,kyn-h-3)和δh7.27(1h,d,j=7.2hz,kyn-h-4)为三重峰,提示存在一个邻位取代的苯环。另外,根据kyn-hβa、kyn-hβb和kyn-h-2同羰基(δc199.0)在hmbc谱中的相关信号,确定该氨基酸为犬尿氨酸。
7个氨基酸残基的连接顺序,是通过分析hmbc和roesy相关信号及esi-ms/ms来确定的。hmbc相关信号arg-nh/kyn-co、kyn-nh/phe-co、phe-nh/pro1-co和val-nh/pro3-co确定了结构片段pro1-phe-kyn-arg和pro3-val的存在。结合roesy相关信号arg-hα/pro2-hδ、pro2-hα/pro3-hα和val-hα/pro1-hδ,确定该环肽的结构为cyclo-(pro1-phe-kyn-arg-pro2-pro3-val)。通过esi-qtof-ms/ms质谱分析发现,b系碎片离子峰:m/z728、538、391、294和195,对应母离子依次丢失arg、kyn、phe、pro和val这些中性分子的碎片离子峰;相应地,y系碎片离子峰:m/z787、690、591、494、347和157,对应母离子依次丢失pro、pro、val、pro、phe和kyn,这证实了该环肽的nmr结构解析结果(图2)。
各氨基酸残基的绝对构型通过高级marfey’s法确定(图3)。本发明化合物(0.1mg)用6n的盐酸在110℃下水解6h后用l-fdla衍生化,相应的l-氨基酸标准品用d/l-fdla衍生化,得到的衍生物用uplc-esi-qtof-ms分析,通过比较样品中各个氨基酸衍生物同标准品衍生物的保留时间来确定绝对构型。氨基酸标准品衍生物的保留时间如下:l-fdla-l-pro14.33min,d-fdla-l-pro15.95min;d-fdla-l-arg9.53min,l-fdla-l-arg10.03min;l-fdla-l-kyn16.67min,d-fdla-l-kyn18.01min;l-fdla-l-phe17.24min,d-fdla-l-phe18.71min;l-fdla-l-val16.06min,d-fdla-l-val18.28min。分析结果表明,该环七肽中的所有氨基酸残基均为l-构型。
实施例2通过合成制备本发明化合物1
化合物1的合成路线
合成步骤如下:
将第一种氨基酸装载到2-ctc树脂中:将2-ctc树脂(100mg,上样量:1.0mmol/g)在装有2ml无水dcm的一次性容器(torivq)中溶胀20分钟。加入fmoc-pro-oh(2.0当量)和diea(4.0当量)的dcm溶液,并将反应容器在室温下在涡旋中摇动1小时。向反应混合物中加入200μlmeoh,并将树脂旋转15分钟。过滤树脂并用无水dcm(3ml×5次,1分钟/次),1:1dcm/meoh(v/v)(3ml×5次,1分钟/次)和meoh(3ml×2次,1分钟/次)洗涤。
fmoc脱保护:在室温下,使用3ml含20%哌啶的dmf溶液进行fmoc脱保护20分钟,然后将树脂用dmf洗涤(3ml×2次,1分钟/次)。
肽偶联:将所需的每种fmoc氨基酸(3个当量),hatu(3个当量)和diea(6个当量)在dmf中室温下与树脂轻轻涡旋搅拌1小时,然后将树脂用dmf洗涤(3ml×5次,1分钟/次)。
fmoc脱保护:在室温下,使用3ml含20%哌啶的dmf溶液进行fmoc脱保护20分钟,然后将树脂用dmf洗涤(3ml×2次,1分钟/次)。
裂解:过滤树脂,并用3ml20%(v/v)hfip裂解溶液在无水dcm中处理1小时,然后重复此步骤处理30分钟。过滤后,合并所得裂解溶液,真空浓缩,通过hplc纯化,得到侧链保护的线性肽1a(8.7mg)。
大环化:将线性肽1a(1当量)在dcm中的溶液加入edci(3当量),hoat(3当量)和diea(3当量),将该溶液在室温搅拌16h,然后真空除去溶剂。
侧链脱保护:向含保护基的环肽中加入1ml裂解混合物(tfa:tis:h2o=95/2.5/2.5,v/v/v),搅拌3h,通过lc-ms监测反应。将该溶液真空浓缩,然后通过hplc纯化,得到前体环肽1b(4.8mg,收率55%)。
臭氧氧化:将前体肽1b(4.8mg,0.005mmol)溶于0.5mlmeoh中,冷却至-78℃,使用臭氧发生器将o3鼓入反应5分钟。在-78℃下将二甲基硫醚(40μl)添加到反应中,使反应混合物在1小时内升温至室温。将反应混合物在真空下浓缩,然后溶于0.5ml的tfa/h2o(1/1,v/v)的混合物中,并在室温下搅拌1h得到本发明化合物4.6mg,历经3步产率95%。
通过比较lc-ms/ms和nmr数据,证明合成的本发明化合物1同天然发现的完全匹配(图4-6)。
实施例3本发明化合物1的体外活性实验
细胞毒活性实验:本发明化合物1测试了对六株肿瘤细胞(mcf-7、hela、nci-h460、sw480、pc9和hepg2)和一株正常细胞(h9c2)的细胞毒活性。样品用dmso溶解,低温保存,dmso在最终体系中的浓度控制在不影响检测活性的范围之内,倍比稀释为1-100μg/ml的工作浓度。取对数生长期细胞,制成单细胞悬液1×106个/ml,将该悬液加到96孔板中,每孔加入100μl。于5%co2,37℃培养箱中培养24h后,分别加入各浓度的受试药物,使其终浓度分别为100、50、25、12.5、6.25μg/ml,每个样品均设3个复孔,阴性对照为等体积培养基及相应的dmso浓度为溶媒对照,以消除dmso对细胞生长的影响,阳性对照药为顺铂。于5%co2,37℃培养箱中培养72h后,每孔加入10μlckk8溶液,继续培养4h后,在450nm处测定各孔吸光值(od值)。通过运用线性回归方法确定抑制百分数,进而计算出ic50值。一般情况下,每个样品在测试中均设置复孔(n=3),在结果中以标准偏差(sd)表示。化合物1对不同细胞半数有效抑制浓度ic50值如表2所示。
表2:化合物1的细胞毒活性
由表2可见,化合物1对mcf-7、hela、nci-h460和pc9这四株肿瘤细胞具有细胞毒作用,ic50值分别为3.4、6.2、7.1和12μm,与阳性药顺铂药效相当,对正常细胞h9c2毒性很低ic50>100μm。本发明化合物是潜在抗肿瘤药物,为研制新的抗肿瘤药物提供了新的先导化合物。
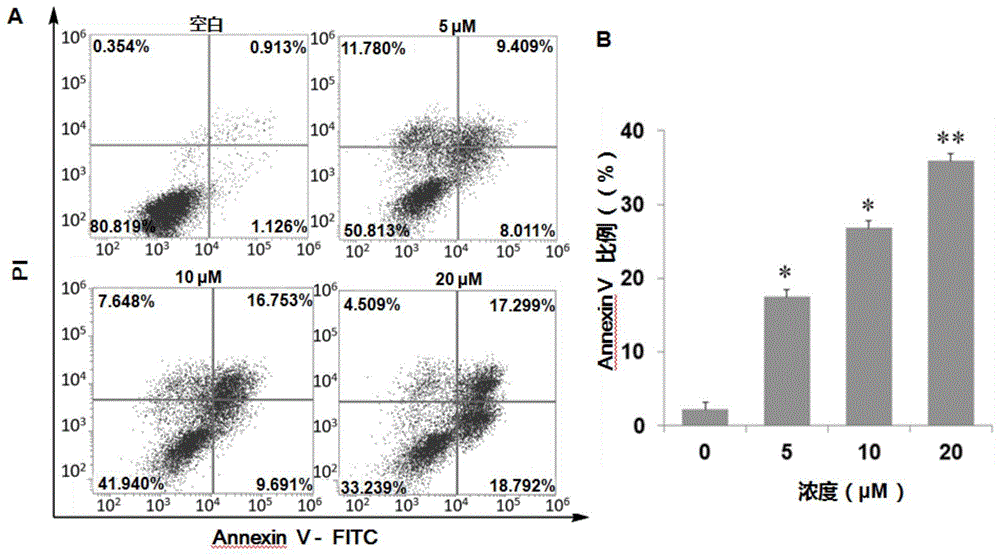
流式细胞技术检测凋亡:取对数生长期的hela细胞,使用0.25%胰酶进行消化处理,获得单细胞悬液,吹打均匀后计数,以1×106个/孔的密度接种至6孔板中,每孔2ml的细胞悬液,十字交叉混匀,置于37℃,5%co2恒温培养箱中培养24h。实验组加入受试样品溶液(5、10、20μm),空白组加入dmso(0.1%),置于37℃,5%co2恒温培养箱中培养48h。胰酶消化,收集细胞,转移到离心管内离心(1000rpm,5min)弃上清。加入500μlbindingsolution轻轻重悬细胞,制成1×106cells/ml的细胞悬液。加入5μlannexinv和fitc结合物,轻轻混匀后室温避光孵育15min;再加入5μlpisolution,室温避光孵育15min,然后用流式细胞仪进行检测,其中annexinv,fitc为第一通路检测,pi为第二通道检测。数据以平均值(mean)±标准偏差(sd)表示,使用spss进行统计学分析,若符合正态分布,多组间比较采用anova分析,组间两两比较采用dunnett’st检验。
从图7的检测结果可以看出本发明化合物1浓度依赖性地诱导hela细胞凋亡。
流式细胞技术检测细胞周期:取稳定生长的hela细胞消化制成单细胞悬液,以1×106/孔的密度接种于6孔板,每孔2ml,37℃,5%co2培养箱中培养过夜。用不同浓度的fuscasine(5、10、20μm)处理24h,胰酶消化后细胞离心(1000rpm,5min)收集细胞。弃去上清,加入4℃预冷过的70%乙醇1ml,放入4℃冰箱固定过夜。离心(1000rpm5min)弃上清,1mlpbs洗涤重悬后离心。弃上清,每管加入500μlpi/rnase,室温孵育15min过滤后,用流式细胞仪检测。数据以平均值(mean)±标准偏差(sd)表示,使用spss进行统计学分析,若符合正态分布,多组间比较采用anova分析,组间两两比较采用dunnett’st检验。本发明化合物1浓度依赖性地诱导hela细胞周期阻滞在g2/m期(图8)。
rxrα双报告基因检测实验:293t细胞接种于10cm细胞培养皿,达到80%融合时转染质粒(pg5luc:pbind-rxra-lbd:lipo2000=10μg:3μg:20μl)。于37℃,5%co2培养箱培养12小时0.25%胰酶进行消化处理,获得单细胞悬液,吹打均匀后计数,以1×104个/孔的密度接种至96孔板中,继续培养12h。实验组9-cis联合20μm的样品处理细胞,对照组用9-cis单独处理细胞,每组做2个复孔,继续培养12h。收集细胞,裂解细胞,细胞上清、萤火虫萤光素酶检测试剂和海肾萤光素酶检测缓冲液于室温平衡。加入萤火虫萤光素酶检测试剂,混匀后于发光检测仪上检测,得到luciferase的读数;加入海肾萤光素酶检测试剂,混匀后于发光检测仪上检测,得到rellina的读数,计算萤火虫荧光素酶的相对活性。
在rxrα双报告基因检测中发现,在样品浓度为20μm时,本发明化合物与9-cis-ra联用能明显抑制9-cis-ra对rxr-α的转录激活作用,提示本发明化合物可能是潜在的与rxr-α相互作用的小分子化合物(图9a)。
westernblot分析:培养3-5代稳定生长的mcf-7和hela细胞,0.25%胰蛋白酶消化后制成悬液,以1×106/孔的密度接种于6孔板,每孔2ml。十字交叉混匀,置于37℃,5%co2恒温培养箱中培养24h。实验组加入受试样品(10、20μm),空白组加dmso(0.1%),置于37℃,5%co2培养箱内培养24h。弃掉上清液,将培养板置于冰上,加入预冷pbs洗涤2次,每孔加入100-200μl细胞裂解液(含pmsf),反复吹打,冰上裂解3min。将细胞碎片及裂解液于4℃12000rpm离心5min,吸取离心管上层蛋白并分装,去除不溶性蛋白裂解液。蛋白浓度使用bca蛋白浓度测定试剂盒测定,量取上清体积,并加入1/5体积的sds-page蛋白上样缓冲液(5×),100℃水浴5min,-20℃保存。蛋白在5%聚丙烯酰胺凝胶sds-page中进行电泳,而后转移到pvdf膜上,将pvdf膜在tbst中漂洗,然后放入5%脱脂奶粉/tbst中,脱色摇床上缓慢摇动,室温封闭1h。弃封闭液,tbst在脱色摇床上洗涤pvdf膜,一抗(1:1000)稀释后4℃冰箱内孵育过夜。回收一抗孵育液,tbst摇床上漂洗pvdf膜,用二抗(1:5000)稀释后室温摇床上慢摇孵育1小时。回收二抗,tbst漂洗。显色剂显色后将pvdf膜置于扫膜仪上,调整参数后扫膜。
如图9所示,本发明化合物1能够明显抑制pi3k催化亚基p85及p-akt的表达,并且能够增加parp蛋白的切割,说明本发明化合物剂量依赖性地抑制mcf-7和hela细胞中akt的活化,且促进凋亡。以上结果提示本发明化合物可能通过rxr-α介导的pi3k/akt信号通路抑制细胞的生长。
分子对接测定。在discoverystudio2.5软件中,绘制本发明化合物的结构作为配体,从proteinbank下载pdb(pdb代码:3nsq)文件作为受体。按照discoverystudio2.5的libdock标准操作流程,使用默认设置将本发明化合物对接到3nsq口袋中进行评分。在discoverystudiovisualizer客户端中可视化本发明化合物的最佳结合形态(图10)。
实施例4~8化合物2-6的合成
本发明参照实施例2的方法合成制得下表3中的化合物2~6。其中化合物5和化合物6为线性肽,无需大环化步骤。
进一步的实验表明化合物2~6对肿瘤细胞也显示出较佳的抑制活性。
表3化合物2~6
- 还没有人留言评论。精彩留言会获得点赞!