制备官能化聚噻吩的方法与流程
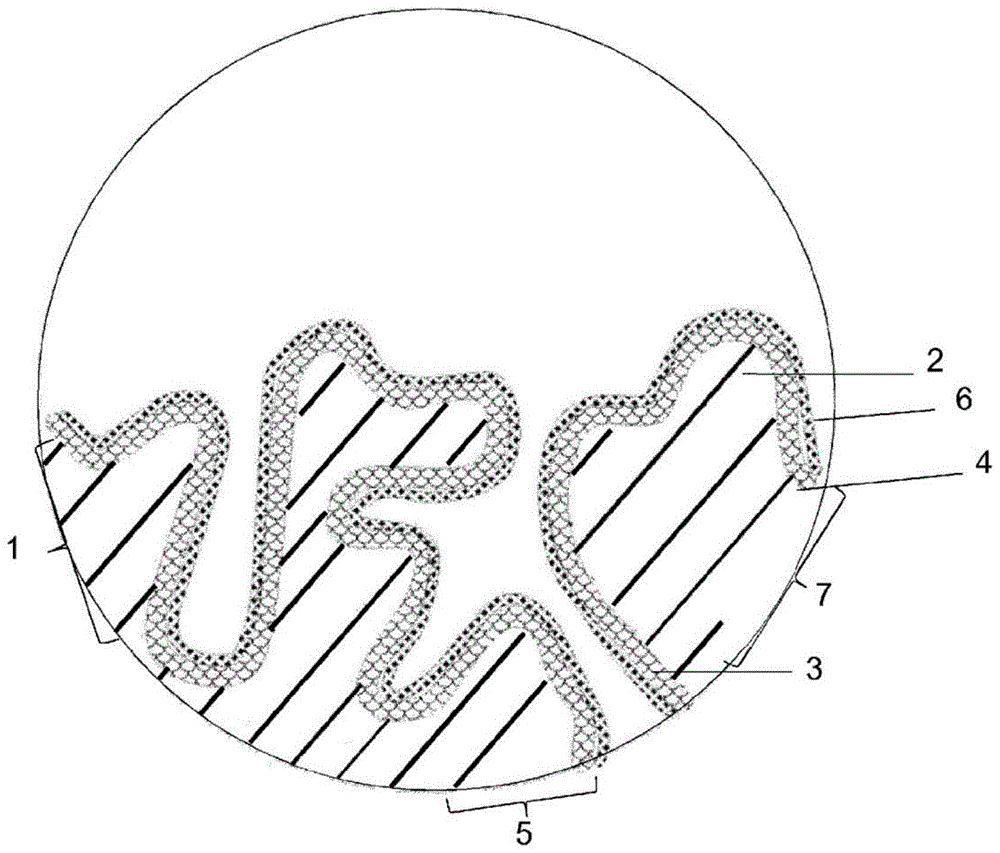
本申请是申请日为2015年11月20日,申请号为201580067823.9,发明名称为“制备官能化聚噻吩的方法”的发明专利申请的分案申请。本发明涉及一种制备包含官能化的π共轭聚噻吩的液体组合物的方法;一种可通过该方法获得的液体组合物;一种包含官能化的π共轭聚噻吩的液体组合物,其中所述组合物的特征在于官能化的π共轭聚噻吩的质均分子量mw与摩尔平均分子量mn的特定比例;一种包含官能化的π共轭聚噻吩的液体组合物,其中官能化的π共轭聚噻吩包含预定量的不同重复单元;一种制备该液体组合物的方法;一种制备电容器的方法,其中这些液体组合物用于形成固体电解质;一种可通过该方法获得的电容器;以及所述液体组合物用于制备导电层的用途。市售的电解电容器通常由多孔金属电极、位于金属表面上的用作电介质的氧化物层、引入多孔结构中的导电材料(通常为固体)、外电极(触点,例如银层)以及进一步的电触点和封装形成。通常使用的电解电容器是钽电解电容器,其阳极由阀金属钽制成,其上通过阳极氧化产生(也称为“形成”)均匀的五氧化二钽介电层。液体或固体电解质形成电容器的阴极。此外,通常使用铝电容器,其中阳极由阀金属铝制成,其上通过阳极氧化产生均匀的电绝缘氧化铝层以作为电介质。此处,液体电解质或固体电解质也形成电容器的阴极。铝电容器通常构造为卷绕或堆叠式电容器。π共轭聚合物由于其高电导率而特别适合作为上述电容器中的固体电解质。π共轭聚合物也称为导电聚合物或合成金属。它们的经济重要性日益增加,因为聚合物在加工性、重量和通过化学改性对性能进行靶向调节方面优于金属。已知的π共轭聚合物的实例为聚吡咯、聚噻吩、聚苯胺、聚乙炔、聚亚苯基和聚(对亚苯基-亚乙烯基),工业上使用的特别重要的聚噻吩为聚(3,4-乙撑二氧噻吩)(pedot),因为其在氧化形式下具有极高的电导率。基于导电聚合物的固体电解质可以以各种方式施加至氧化物层上。ep-a-0340512描述了例如由3,4-乙撑二氧噻吩制备固体电解质及其在电解电容器中的用途。根据该公开文献的教导,3,4-亚乙二氧基噻吩在氧化物层上原位聚合。除原位聚合之外,现有技术还已知一种制备电容器中的固体电解质的方法,其中将包含已聚合的噻吩和作为抗衡离子的聚阴离子的分散体,例如现有技术已知的pedot/pss分散体(pedot=聚(3,4-乙撑二氧噻吩),pss=聚苯乙烯磺酸)施加至氧化物层上,然后通过蒸发移除分散剂。该制备固体电解质电容器的方法例如公开在de-a-102005043828中。然而,pedot/pss分散体的特征在于如下缺点:它们包含显著量的作为非导电惰性材料的pss。此外,由于存在pss,分散体中pedot/pss颗粒的尺寸有时太大,以至于不能确保颗粒也渗入多孔金属电极的较小的孔中。最后,pedot/pss分散体的最大固含量通常被限制在约3重量%的值。为了克服这些缺点,已经制备了包含pedot衍生物的液体组合物,其不具有已知的pedot/pss分散体的缺点。首先开发了用磺酸根官能化的聚噻吩。由于磺酸根,这些聚噻吩是自掺杂的,不需要诸如pss的抗衡离子。例如,ep1122274a1公开了官能化的π共轭聚合物的制备,例如通过氧化聚合相应单体4-(2,3-二氢噻吩并[3,4-b][1,4]二氧杂环己烯(dioxin)-2-基甲氧基)-1-丁磺酸(edot-s)而制备聚(4-(2,3-二氢噻吩并[3,4-b][1,4]二氧杂环己烯-2-基甲氧基)-1-丁磺酸)(pedot-s)。然而,由ep1122274a1中获得的聚合物溶液制备的导电层的电导率通常过低,以至于不能使用这些聚合物溶液来制备例如固体电解质电容器中的固体电解质层。因此,本发明的目的是克服水溶性π共轭聚合物领域中的现有技术的缺点。特别地,本发明的目的是提供包含水溶性或水分散性π共轭聚合物的组合物,与现有技术已知的相应组合物相比,其特征在于由这些组合物制备的导电层的电导率增加。本发明的目的还在于提供包含水溶性或水分散性π共轭聚合物的组合物,其在用于形成电容器中的固体电解质层时,导致电容器的有利性质,特别是与固体电解质层使用现有技术已知的相应组合物制备的电容器相比,有利的电容和/或有利的esr(等效串联电阻)。此外,本发明的目的还在于提供包含水溶性或水分散性π共轭聚合物的组合物,其在用于形成电容器中的固体电解质层时,有助于提高电容器在高温和高相对湿度下储存时的稳定性。电容器在该存储条件下的稳定性是根据电容器的电性能,特别是电容和esr在存储期间的劣化程度定义的。对实现至少一个上述目的的贡献由类别形成性独立权利要求的主题提供,其中从属的子权利要求表示本发明的优选实施方案,其主题同样对实现至少一个目的构成贡献。实施方案i.一种制备包含官能化的π共轭聚噻吩的液体组合物的方法,所述方法包括如下步骤:i)提供包含如下的液相:a)通式(i)的噻吩单体:其中:x,y相同或不同,为o、s或nr1,其中r1为氢或具有1-18个碳原子的脂族或芳族残基;a为带有阴离子官能团的有机残基;b)氧化剂;和c)溶剂;ii)使通式(i)的噻吩单体氧化聚合,从而获得包含官能化的π共轭聚噻吩的液体组合物;其中:(α1)将工艺步骤i)中提供的液相的ph调节至低于7.0的值,其中ph在20℃的温度下测定;和(α2)工艺步骤i)中提供的液相的氯离子含量基于液相的总重量小于10000ppm。ii.根据实施方案i的方法,其中:(α3)工艺步骤i)中提供的液相的氧含量基于液相的总重量小于1000ppm。iii.根据实施方案i或ii的方法,其中:x,y为o,a为-(ch2)m-cr2r3-(ch2)n-,其中:r2为氢或-(ch2)s-z-(ch2)p-so3-m+,r3为-(ch2)s-z-(ch2)p-so3-m+,z为o、s或-ch2-,m+为阳离子,m和n相同或不同,为0-3的整数,s为0-10的整数,p为1-18的整数。iv.根据前述实施方案中任一项的方法,其中在通式(i)中,x,y为o,a为-(ch2)-cr2r3-(ch2)n-,其中:r2为氢,r3为-(ch2)s-o-(ch2)p-so3-m+,m+为na+或k+,n为0或1,s为0或1,且p为4或5。v.根据实施方案iv的方法,其中:x,y为o,a为-(ch2-chr)-,其中:r为-(ch2)t-o-ar-[(w)u-so3-m+]v,其中:ar表示任选取代的c6-c20亚芳基;w表示任选取代的c1-c6亚烷基;m+表示h+,选自li+、na+和k+的碱金属阳离子,nh(r1)3或hnc5h5,其中各r1基团独立地表示氢原子或任选取代的c1-c6烷基;t表示0-6的整数,u表示0或1的整数;且v表示1-4的整数。vi.根据前述实施方案中任一项的方法,其中氧化剂b)为重金属盐、过二硫酸盐或其混合物。vii.根据实施方案i至v中任一项的方法,其中噻吩单体通过电化学聚合而聚合,且其中氧化剂b)为电极。viii.根据前述实施方案中任一项的方法,其中溶剂c)为水。ix.根据前述实施方案中任一项的方法,其中使用有机或无机酸将工艺步骤i)中提供的液相的ph调节至低于7.0的值。x.根据前述实施方案中任一项的方法,其中工艺步骤ii)中的氧化聚合在氮气、氩气、二氧化碳或其混合物的惰性气体气氛下进行。xi.根据实施方案x的方法,其中工艺步骤ii)中的氧化聚合在等于或高于工艺步骤ii)中的聚合反应期间液相的蒸气压的压力下进行。xii.根据前述实施方案中任一项的方法,其中工艺步骤ii)中的氧化聚合在不大于0.8巴的减压下进行。xiii.根据前述实施方案中任一项的方法,其中在另一工艺步骤iii)中,将工艺步骤ii)中获得的液体组合物纯化。xiv.根据实施方案xiii的方法,其中纯化通过过滤和/或通过用离子交换剂处理实现。xv.根据前述实施方案中任一项的方法,其中通过用超声波处理液体组合物、通过高压均化处理液体组合物或通过热处理液体组合物而调节在工艺步骤ii)或工艺步骤iii)中获得的液体组合物中的官能化π共轭聚噻吩的粒度分布。xvi.一种可通过根据前述实施方案中任一项的方法获得的液体组合物。xvii.一种包含官能化的π共轭聚噻吩的液体组合物,其中所述聚噻吩包含通式(i)的重复单元:其中x、y和a如实施方案i、iii、iv和v所定义,且其中官能化的π共轭聚噻吩的质均分子量mw与摩尔平均分子量mn(mw/mn)之比为至少6,优选至少8,更优选至少10,更优选至少12,更优选至少14,更优选至少16,更优选至少18,更优选至少20。xviii.根据实施方案xvii的液体组合物,其中mw/mn为至多100,优选至多75,更优选至多50。xix.根据实施方案xvii或xviii的液体组合物,其中官能化的π共轭聚噻吩的质均分子量mw为至少50000g/mol,优选至少75000g/mol,更优选至少100000g/mol,最优选至少125000g/mol。xx.根据实施方案xvii至xix中任一项的液体组合物,其中官能化的π共轭聚噻吩的质均分子量mw为125000-240000g/mol,优选为125000-210000g/mol。xxi.根据实施方案xvii至xx中任一项的液体组合物,其中官能化的π共轭聚噻吩的摩尔平均分子量mn小于25000g/mol,优选小于20000g/mol,更优选小于15000g/mol。xxii.根据实施方案xvii至xxi中任一项的液体组合物,其中在液体组合物中,官能化的π共轭聚噻吩以颗粒形式存在,其中粒度分布的特征在于:i)d50值(重均粒径)为1-100nm,优选为1-80nm,更优选为1-60nm,最优选为5-40nm,和ii)d90值小于3.5×d50,优选小于3×d50,更优选小于2×d50。xxiii.根据实施方案xvii至xxii中任一项的液体组合物,其中官能化的π共轭聚噻吩包含通式(ia)的重复单元和通式(ib)的重复单元:并且其中通式(ib)重复单元的含量小于20重量%,优选小于18重量%,更优选小于16重量%,更优选小于14重量%,更优选小于12重量%,最优选小于10重量%,并且通式(ia)重复单元的含量大于80重量%,更优选大于82重量%,更优选大于84重量%,更优选大于86重量%,更优选大于88重量%,最优选至少90重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量,其中通式(ia)重复单元的含量和通式(ib)重复单元的含量优选总计为100重量%。xxiv.根据实施方案xxiii的液体组合物,其中通式(ib)重复单元的含量为至少0.2重量%,优选至少1重量%,更优选至少2重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量。xxv.一种制备包含官能化的π共轭聚噻吩的液体组合物的方法,所述方法包括如下步骤:i)提供包含如下的液相:a)通式(i)的噻吩单体:其中x、y和a如实施方案i、iii、iv和v所定义,其中液相包含通式(ia)的噻吩单体和通式(ib)的噻吩单体的混合物:并且其中通式(ib)的噻吩单体的含量小于20重量%,优选小于18重量%,更优选小于16重量%,更优选小于14重量%,更优选小于12重量%,最优选小于10重量%,并且通式(ia)的噻吩单体的含量大于80重量%,更优选大于82重量%,更优选大于84重量%,更优选大于86重量%,更优选大于88重量%,最优选大于90重量%,在每种情况下基于液相中的噻吩单体总重量,其中通式(ia)的噻吩单体的含量和通式(ib)的噻吩单体的含量优选总计为100重量%;a)氧化剂;和b)溶剂;ii)将通式(ia)和(ib)的噻吩单体氧化聚合,从而获得包含官能化的π共轭聚噻吩的液体组合物。xxviii.根据实施方案xxv的方法,其中通式(ib)的噻吩单体的含量为至少0.2重量%,优选至少1重量%,更优选至少2重量%,在每种情况下基于液相中的噻吩单体总重量。xxvii.可通过根据实施方案xxv或xxvi的方法获得的液体组合物。xxviii.一种包含官能化的π共轭聚噻吩的液体组合物,其中所述聚噻吩包含通式(i)的重复单元:其中x、y和a如实施方案i、iii、iv和v所定义,其中官能化的π共轭聚噻吩包含通式(ia)的重复单元和通式(ib)的重复单元:并且其中通式(ib)的重复单元的含量小于20重量%,优选小于18重量%,更优选小于16重量%,更优选小于14重量%,更优选小于12重量%,最优选小于10重量%,并且通式(ia)的重复单元的含量大于80重量%,更优选大于82重量%,更优选大于84重量%,更优选大于86重量%,更优选大于88重量%,最优选大于90重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量,其中通式(ia)的重复单元的含量和通式(ib)的重复单元的含量优选总计为100重量%。xxix.根据实施方案xxviii的液体组合物,其中通式(ib)的重复单元的含量为至少0.2重量%,优选至少1重量%,更优选至少2重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量。xxx.根据实施方案xvi至xxiv和xxvii至xxix中任一项的液体组合物,其中由所述液体组合物制备的导电层的电导率大于12s/cm。xxxi.一种制备电容器的方法,包括如下工艺步骤:i)提供电极材料的电极体,其中电介质在形成阳极体下至少部分覆盖该电极材料的一个表面;ii)将根据实施方案xvi至xxiv和xxvii至xxx中任一项的液体组合物引入电极体的至少一部分中。xxxii.一种可通过根据实施方案xxxi的方法获得的电容器。xxxiii.根据实施方案xvi至xxiv和xxvii至xxx中任一项的液体组合物用于制备电子器件中的导电层的用途。xxxiv.根据实施方案xxxiii的用途,其中所述器件选自光导电池、光敏电阻器、光开关、光敏晶体管、光电管、ir检测器、光伏器件、太阳能电池、用于存储器件的涂料材料、场效应电阻器件、抗静电膜、生物传感器、电致变色器件、固体电解质电容器、储能器件、触摸屏和电磁屏蔽。xxxv.根据实施方案xxxiii的用途,其中所述导电层为固体电解质电容器中的固体电解质层。对实现这些目的的贡献由用于制备包含官能化的π共轭聚噻吩的液体组合物的第一方法做出,所述方法包括如下步骤:i)提供包含如下的液相:a)通式(i)的噻吩单体:其中:x,y相同或不同,为o、s或nr1,其中r1为氢或具有1-18个碳原子的脂族或芳族残基;a为带有阴离子官能团的有机残基,优选选自-co2-、-so3-和-oso3-的阴离子官能团,其中特别优选-so3-;b)氧化剂;和c)溶剂;ii)使通式(i)的噻吩单体氧化聚合,从而获得包含官能化的π共轭聚噻吩的液体组合物;其中:(α1)将工艺步骤i)中提供的液相的ph调节至低于7.0,优选低于6.0,更优选低于5.0,更优选低于4.0,更优选低于3.0,更优选低于2.0,最优选低于1.0的值,其中ph在20℃的温度下测定;和(α2)工艺步骤i)中提供的液相的氯离子含量小于10000ppm,更优选小于5000ppm,更优选小于1000ppm,更优选小于500ppm,最优选小于100ppm,在每种情况下基于水相的总重量。令人惊讶地发现,可根据ep1122274a1中公开的方法通过相应的单体的氧化聚合来制备包含能够形成具有提高的导电性的导电层的官能化π共轭聚噻吩(如pedot-s)的液体组合物,条件是在聚合反应之前,将单体溶液的ph调节至低于7.0的值,并保持该单体溶液中的氯含量低于10000ppm。在本发明第一方法的工艺步骤i)中,提供了包含噻吩单体a)、氧化剂b)和溶剂c)的液相。根据本发明第一方法的第一实施方案,噻吩单体a)为在ep1122274a1中公开的那些。根据这些噻吩单体a)的优选实施方案,通式(i)中的x和y均为氧(o),其中特别优选:a为-(ch2)m-cr2r3-(ch2)n-,其中:r2为氢或-(ch2)s-o-(ch2)p-so3-m+,r3为-(ch2)s-o-(ch2)p-so3-m+,m+为阳离子,优选为h+、li+、na+、k+、rb+、cs+或nh4+,特别优选为na+或k+,m和n相同或不同,且为0-3的整数,优选为0或1,s为0-10的整数,优选为0或1,且p为1-18的整数,优选为4或5。在本文中,甚至更优选:a为-(ch2)-cr2r3-(ch2)n-,其中:r2为氢,r3为-(ch2)s-o-(ch2)p-so3-m+,m+为na+或k+,n为0或1,s为0或1,且p为4或5。与本发明方法的第一实施方案相关的最优选的官能化π共轭聚噻吩为聚(4-(2,3-二氢噻吩并[3,4-b][1,4]二氧杂环己烯-2-基甲氧基)-丁磺酸)(pedot-s);因此,最优选的噻吩单体a)为4-(2,3-二氢噻吩并[3,4-b][1,4]二氧杂环己烯-2-基甲氧基)-1-丁磺酸(edot-s)。然而,用于制备pedot-s的edot-s单体可包含一定量的如ep1564250a1中所公开的prodot-s(4-(3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环庚烯-3-基)-1-丁磺酸)。根据本发明第一方法的第二实施方案,噻吩单体a)为jp2014-028760a中公开的那些。因此,在本文中,优选在通式(i)中:x,y为o,a为-(ch2-chr)-,其中:r为-(ch2)t-o-ar-[(w)u-so3-m+]v,其中:ar表示任选取代的c6-c20亚芳基;w表示任选取代的c1-c6亚烷基;m+表示h+,选自li+、na+和k+的碱金属阳离子,nh(r1)3或hnc5h5,其中各r1基团独立地表示氢原子或任选取代的c1-c6烷基;t表示0-6的整数,u表示0或1的整数;且v表示1-4的整数。在本文中,特别优选的噻吩单体为jp2014-028760a第[0049]段中明确提及的那些。在工艺步骤ii)中进行的氧化反应可通过化学氧化剂、电化学氧化或这两种方法的组合来催化。在电化学氧化的情况下,电极起氧化剂b)的作用。适于用作化学氧化剂的氧化剂b)为重金属盐,优选铁盐,更优选fecl3以及芳族和脂族磺酸的铁(iii)盐,h2o2,k2cr2o7,过二硫酸盐如k2s2o8、na2s2o8,kmno4,碱金属过硼酸盐和碱金属过硫酸盐或过硫酸铵,或这些氧化剂的混合物。特别优选重金属的盐、过二硫酸盐或其混合物。其他合适的氧化剂例如描述于handbookofconductingpolymers(编辑skotheim,t.a.),marceldekker:newyork,1986,第1卷第46-57页中。特别优选的氧化剂b)为过二硫酸盐,特别为k2s2o8、na2s2o8,铁盐,特别是氯化铁(iii),或过二硫酸盐与至少一种催化过二硫酸盐分解的其他化合物的混合物,例如过二硫酸盐与铁盐的混合物。然而,鉴于要求(α2),即工艺步骤i)中提供的液相的氯离子含量小于10000ppm,不包含任何氯离子或者以使得仍满足要求(α2)的低含量包含氯离子的那些氧化剂b)是优选的。根据本发明方法特别优选的实施方案,氧化剂为fe2(so4)3和na2s2o8的混合物。可用于本发明第一方法中的合适溶剂c)为水,水混溶性溶剂,特别是选自如下的那些:脂族醇如甲醇、乙醇、异丙醇和丁醇,双丙酮醇,乙二醇和甘油,脂族酮如丙酮和甲基乙基酮,脂族亚硝酸盐如乙腈,或这些溶剂中至少两种的混合物,特别是水和水混溶性溶剂的混合物。然而,最优选的溶剂为水。因此,在使用4-(2,3-二氢噻吩并[3,4-b][1,4]二氧杂环己烯-2-基甲氧基)-1-丁磺酸(edot-s)作为噻吩单体a)的情况下,本发明的第一方法能制备pedot-s的水溶液。工艺步骤i)中提供的水相中的噻吩单体a)浓度优选为0.1-25重量%,优选为0.5-10重量%。存在不同的方式来制备工艺步骤i)中提供的液相。可将噻吩单体a)溶解或分散在溶剂c)中,然后加入氧化剂b)(其也可单独溶解或分散在溶剂中);或者将氧化剂b)首先溶解或分散在溶剂c)中,然后加入噻吩单体a)(其也可单独溶解或分散在溶剂中)。此外,如果使用多于一种氧化剂,如fe2(so4)3和na2s2o8的混合物,则可首先将这些组分中的一种与噻吩单体a)和溶剂c)混合,最后加入第二种氧化剂。与工艺步骤i)中的液相制备方式无关,特别优选将用于制备液相的组分中的氧含量降至液相中的氧含量小于1000ppm,更优选小于500ppm,更优选小于100ppm,更优选小于10ppm,更优选小于1ppm,更优选小于0.5ppm,最优选小于0.25ppm的程度,在每种情况下基于液相的总重量。根据本发明方法的特别优选的实施方案,用于制备液相的组分完全不含任何氧(即氧含量为0ppm)。氧含量的降低可例如通过在减压下搅拌用于制备液相的组分、通过使用超声波或通过使用惰性气体如n2、氩气、co2或其混合物将这些组分脱气,或通过上述方法的组合实现。工艺步骤ii)中的聚合反应优选在-20℃至200℃,优选0-100℃的温度下进行,持续时间优选为1-48小时,更优选为5-20小时。在聚合反应结束后,包含官能化的π共轭聚合物的液体组合物,优选pedot-s水溶液可进一步纯化,例如通过过滤,特别是通过超滤,和/或通过在另一工艺步骤iii)中用离子交换剂处理,特别是通过用阴离子交换剂和阳离子交换剂处理,以进一步纯化。如下文就制备电容器的方法所述的那样,还可添加其他添加剂。此外,由于在工艺步骤ii)中聚合后获得的官能化π共轭聚噻吩通常以颗粒形式存在,因此在工艺步骤ii)中获得或者在进一步纯化后在工艺步骤iii)中获得的液体组合物中的官能化π共轭聚噻吩的粒度分布可通过用超声波处理液体组合物来调节,其中能量输入优选为10-1000瓦/升(w/l),更优选为20-500w/l,超声频率优选为20-200khz;通过用高压均化处理液体组合物来调节,其中优选多次施加高于100巴,优选高于500巴,更优选高于1500巴,更优选高于2500巴,更优选高于3500巴,最优选高于4500巴的压力;或者通过热处理液体组合物,其中热处理优选包括在40-100℃,优选50-95℃的温度下处理液体组合物达5分钟至100小时,优选1-10小时,更优选2-8小时的时间。现在,本发明第一方法的特征在于:(α1)将工艺步骤i)中提供的液相的ph调节至低于7.0,优选低于6.0,更优选低于5.0,更优选低于4.0,更优选低于3.0,更优选低于2.0的值,最优选低于1.0的值,其中ph在20℃的温度下测定;和(α2)工艺步骤i)中提供的液相的氯离子含量小于10000ppm,优选小于5000ppm,更优选小于1000ppm,更优选小于500ppm,最优选小于100ppm,在每种情况下基于水相的总重量。根据要求(α1)中的定义将ph值调节至低于7.0的值—鉴于(α2)中定义的要求—优选使用无机或有机酸,优选基本上不含氯离子的有机或无机酸实现。合适的有机酸包括羧酸如甲酸、乙酸、乳酸、丙酸、柠檬酸、苹果酸、富马酸或其混合物。合适的无机酸特别为硫酸、磺酸、硝酸、膦酸、磷酸或其混合物。还可与这些不含氯离子的有机或无机酸中的一种组合地使用含氯离子的辅酸(co-acid),例如盐酸,只要这些辅酸以使得仍满足要求(α2)的低量使用。根据本发明方法的特别优选的实施方案,使用硫酸来调节ph。优选地,通过选择组分a)、b)和c),特别是通过选择基本上不含氯离子的氧化剂b)来将工艺步骤i)中提供的液相的氯离子含量调节至根据要求(α2)所定义的小于10000ppm。需要的话,用于制备液相的组分的氯离子含量可额外通过用阴离子交换剂处理这些组分来降低。根据本发明第一方法的特别优选的实施方案,还有利的是:(α3)工艺步骤i)中提供的液相的氧含量小于1000ppm,优选小于500ppm,更优选小于100ppm,更优选小于10ppm,更优选小于1ppm,更优选小于0.5ppm,最优选小于0.25ppm,在每种情况下基于液相的总重量。根据本发明方法的特别优选的实施方案,工艺步骤i)中提供的液相的氧含量完全不含任何氧(即氧含量为0ppm)。存在不同的方法来调节工艺步骤i)中提供的液相中的氧含量,并且还在工艺步骤ii)中的聚合反应期间保持该低氧含量。根据一种方法,可将工艺步骤i)中提供的液相(或者用于制备液相的液体组分)脱气,例如通过将惰性气体如n2、氩气、co2或其混合物引入在工艺步骤i)中提供的液相中,从而降低液相中的初始氧含量。或者,可对工艺步骤i)中提供的液相(或者用于制备液相的液体组分)进行减压处理以降低初始氧含量,例如通过在施加真空下搅拌液体,或者可用超声波进行处理,或者可实施减压处理和超声处理的组合。为了确保在工艺步骤ii)的聚合反应期间保持低氧含量,可有利的是在惰性气体气氛下,优选在n2气氛下、在co2气氛下、在氩气气氛下或者在这些惰性气体中至少两种的混合物气氛下实施聚合反应,其中还可有利的是,工艺步骤ii)中的氧化聚合在等于或高于液相在工艺步骤ii)中的聚合反应期间的蒸气压的压力下进行。优选地,工艺步骤ii)中的氧化聚合在比工艺步骤ii)中的聚合反应期间的蒸汽压高至少0.1毫巴,更优选至少0.5毫巴,最优选至少1毫巴的压力下进行。为了确保在工艺步骤ii)中的聚合反应期间保持低氧含量,还可在减压下,优选在不超过0.8巴的压力下,最优选在不超过0.5巴的压力下实施工艺步骤ii)中的氧化聚合。对实现上述目的的贡献还由一种包含官能化的π共轭聚噻吩的液体组合物做出,优选由一种可通过本发明第一方法获得的pedot-s水溶液做出,优选由一种包含官能化的π共轭聚噻吩的液体组合物做出,特别是由一种可通过本发明第一方法获得的pedot-s水溶液做出。对实现上述目的的贡献还由一种包含官能化的π共轭聚噻吩的第一液体组合物做出,其中聚噻吩包含通式(i)的重复单元:其中x、y和a如本发明第一方法所定义,并且其中官能化的π共轭聚噻吩的质均分子量mw与摩尔平均分子量mn(mw/mn)之比为至少6,优选至少8,更优选至少10,更优选至少12,更优选至少14,更优选至少16,更优选至少18,更优选至少20。本发明第一液体组合物中所含的官能化π共轭聚噻吩中(以及如后文所述的本发明第二液体组合物中所含的官能化π共轭聚噻吩中)的通式(i)重复单元如下式(i')所示地彼此键合,其中星号(*)表示与相邻重复单元的键合。优选地,官能化的π共轭聚噻吩具有沿聚合物链的正电荷(式(i')中未示出),并且这些正电荷至少部分地由有机残基a中的阴离子官能团补偿。令人惊讶地发现,至少为6的mw/mn值显著地有助于改善官能化的π共轭聚噻吩的性质,特别是如果使用该官能化的π共轭聚噻吩来形成固体电解质电容器中的固体电解质。如果使用mw/mn值至少为6的官能化的π共轭聚噻吩如pedot-s,则与其中使用mw/mn值约4.4的pedot-s来制备固体电解质层的固体电解质电容器(mw=123000g/mol且mn=28000g/mol的该类pedot-s例如公开在“pedotprinciplesanapplicationsofanintrinsicallyconductivepolymer”,elschner等(2011),crc第12.4章中)相比,可获得具有改善的电容和改善的esr的固体电解质电容器。根据本发明液体组合物的优选实施方案,mw/mn的值为至多100,优选为至多75,更优选为至多50。此外优选的是,官能化的π共轭聚噻吩的质均分子量mw为至少50000g/mol,优选为至少75000g/mol,更优选为至少100000g/mol,最优选为至少125000g/mol。此外已表明有利的是,官能化的π共轭聚噻吩的质均分子量mw为125000-240000g/mol,优选为125000g/mol-210000g/mol。此外还优选的是,官能化的π共轭聚噻吩的摩尔平均分子量mn小于25000g/mol,优选小于20000g/mol,更优选小于15000g/mol。如上所述,官能化的π共轭聚噻吩—特别是当通过本发明第一方法制备时—通常以颗粒形式存在。在本文中,特别优选这些颗粒的粒度分布的特征在于:i)d50值(重均粒径)为1-100nm,优选为1-80nm,更优选为1-60nm,最优选为5-40nm,和ii)d90值小于3.5×d50,优选小于3×d50,更优选小于2×d50。就包含具有上文所定义的mw、mn和mw/mn值的官能化π共轭聚噻吩的本发明第一液体组合物而言,此外优选的是,官能化的π共轭聚噻吩包含通式(ia)和通式(ib)的重复单元:其中通式(ib)的重复单元的含量小于20重量%,优选小于18重量%,更优选小于16重量%,更优选小于14重量%,更优选小于12重量%,最优选小于10重量%,并且通式(ia)的重复单元的含量大于80重量%,更优选大于82重量%,更优选大于84重量%,更优选大于86重量%,更优选大于88重量%,最优选大于90重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量,其中优选通式(ib)重复单元的含量和通式(ia)重复单元的含量总计为100重量%(即,官能化的π共轭聚噻吩仅由这两种重复单元构成)。在本文中,此外优选的是,通式(ib)的重复单元的含量为至少0.2重量%,优选为至少1重量%,更优选为至少2重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量。官能化的π共轭聚噻吩中的通式(ia)和(ib)的重复单元的含量相对量可使用包含适当相对量的相应单体的液相并根据本发明的方法氧化聚合这些单体而调节。对实现上述目的的贡献还由一种制备包含官能化的π共轭聚噻吩的液体组合物的第二方法做出,所述方法包括如下步骤:i)提供包含如下的液相:a)通式(i)的噻吩单体:其中x、y和a如上文所定义,其中液相包含通式(ia)的噻吩单体和通式(ib)的噻吩单体的混合物:并且其中通式(ib)的噻吩单体的含量小于20重量%,优选小于18重量%,更优选小于16重量%,更优选小于14重量%,更优选小于12重量%,最优选小于10重量%,并且通式(ia)的噻吩单体的含量大于80重量%,更优选大于82重量%,更优选大于84重量%,更优选大于86重量%,更优选大于88重量%,最优选大于90重量%,在每种情况下基于液相中的噻吩单体总重量,其中通式(ia)的噻吩单体的含量和通式(ib)的噻吩单体的含量优选总计为100重量%;b)氧化剂;和c)溶剂;ii)将通式(ia)和(ib)的噻吩单体氧化聚合,从而获得包含官能化的π共轭聚噻吩的液体组合物。还令人惊讶地发现,如果使用通过包含小于20重量%的通式(ib)噻吩单体和大于80重量%通式(ia)噻吩单体制备的官能化π共轭聚噻吩来形成固体电解质电容器中的固体电解质层,则电容器就电容和esr而言的性能可显著改善。如上所述,用于制备pedot-s的edot-s单体(通式(ia)的重复单元)可包含一定量ep1564250a1中所公开的prodot-s(通式(ib)的重复单元)。已表明将prodot-s的含量调节至小于20重量%的值,优选调节至0.1重量%至不大于14重量%的值,更优选调节至1重量%至不大于12重量%的值,甚至更优选调节至2重量%至不大于10重量%的值是特别有利的,如果使用该官能化的π共轭聚噻吩来形成固体电解质电容器中的固体电解质层。在本文中,还优选通式(ib)的噻吩单体的含量为至少0.2重量%,优选为至少1重量%,更优选为至少2重量%,在每种情况下基于液相中的噻吩单体总重量。优选的溶剂和氧化剂是就本发明第一方法所述的那些。对实现上述目的的贡献还由一种包含官能化的π共轭聚噻吩的液体组合物做出,优选由一种可通过本发明第二方法获得的pedot-s水溶液做出,特别是由通过本发明第二方法获得的pedot-s水溶液做出。对实现上述目的的贡献还由一种包含官能化的π共轭聚噻吩的第二液体组合物做出,其中聚噻吩包含通式(i)的重复单元:其中x、y和a如本发明第一方法所定义,其中官能化的π共轭聚噻吩包含通式(ia)的重复单元和通式(ib)的重复单元:并且其中通式(ib)的重复单元的含量小于20重量%,优选小于18重量%,更优选小于16重量%,更优选小于14重量%,更优选小于12重量%,最优选小于10重量%,并且通式(ia)的重复单元的含量大于80重量%,更优选大于82重量%,更优选大于84重量%,更优选大于86重量%,更优选大于88重量%,最优选大于90重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量,其中通式(ib)重复单元的含量和通式(ia)重复单元的含量优选总计为100重量%(即,官能化的π共轭聚噻吩仅由这两种重复单元构成)。在本文中,还优选通式(ib)的重复单元的含量为至少0.2重量%,优选为至少1重量%,更优选为至少2重量%,在每种情况下基于官能化的π共轭聚噻吩的总重量。在本文中,特别优选的是,由可通过本发明第一或第二方法获得的液体组合物、包含具有非常确定的mw/mn值的官能化π共轭聚噻吩的本发明第一液体组合物,和包含含非常确定的通式(ia)和(ib)重复单元的相对量的官能化π共轭聚噻吩的本发明第二液体组合物制备的导电层具有大于12s/cm,优选大于14s/cm,更优选大于16s/cm,优选大于18s/cm,更优选大于20s/cm,更优选大于25s/cm,更优选大于40s/cm,更优选大于60s/cm,最优选大于80s/cm的电导率。对实现上述目的的贡献还由一种制备电容器的方法做出,其包括如下工艺步骤:i)提供电极材料的电极体,其中电介质在形成阳极体下至少部分覆盖该电极材料的一个表面;ii)将可通过本发明第一或第二方法获得的液体组合物、包含具有非常确定的mw/mn值的官能化π共轭聚噻吩的第一液体组合物,或者包含含非常确定的通式(ia)和(ib)重复单元的相对量的官能化π共轭聚噻吩的本发明第二液体组合物引入电极体的至少一部分中。在工艺步骤i)中,首先提供电极材料的电极体,其中电介质至少部分覆盖该电极材料的一个表面,从而形成阳极体。原则上,电极体可通过压制具有高表面积的阀金属粉末并将其烧结以得到通常为多孔的电极体而制备。此时,通常也将电接触线(优选为阀金属,例如钽)压入电极体中。然后,用电介质(即氧化物层)涂覆电极体,例如通过电化学氧化。或者,也可蚀刻金属箔,并通过电化学氧化用电介质涂覆,从而获得具有多孔区域的阳极箔。在卷绕式电容器中,将形成电极体的具有多孔区域的阳极箔和阴极箔由隔膜隔开并卷绕。在本发明的上下文中,阀金属应理解为意指其氧化物层不能使电流在两个方向上相等流动的那些金属。在阳极施加电压的情况下,阀金属的氧化物层阻挡电流,而在阴极施加电压的情况下,会产生大电流,这可能会破坏氧化物层。阀金属包括be、mg、al、ge、si、sn、sb、bi、ti、zr、hf、v、nb、ta和w以及这些金属中的至少一种与其他元素的合金或化合物。最为人所知的代表性阀金属为al、ta和nb。具有与阀金属相当的电性能的化合物为具有金属导电性的那些,其可以被氧化,并且其氧化物层具有上述性质。例如,nbo具有金属导电性,但通常不被认为是阀金属。然而,氧化的nbo层具有阀金属氧化物层的典型性质,因此nbo或nbo与其他元素的合金或化合物是具有与阀金属相当的电性能的该类化合物的典型实例。优选钽,铝的电极材料和基于铌或氧化铌的那些电极材料。作为电极材料,非常特别优选钽和铝。为了制备通常具有多孔区域的电极体,可将阀金属(例如呈粉末形式)烧结,从而获得通常为多孔的电极体,或者将多孔结构压印至金属体上。后者可例如通过蚀刻箔进行。简单起见,具有多孔区域的物体在下文中也称为多孔的。因此,例如,具有多孔区域的电极体也称为多孔电极体。一方面,多孔体可被多个通道穿透,因此是海绵状的。如果使用钽来构建电容器,则通常存在这种情况。此外,可能只有表面才具有孔,并且表面孔下方的区域就结构而言是实心的。如果使用铝来构建电容器,则通常会观察到这种情况。优选地,电极体是多孔的。然后,将以此方式制得的通常为多孔的电极体例如在合适的电解质,例如磷酸或己二酸铵水溶液中通过施加电压而氧化,从而形成电介质。该形成电压的水平取决于待获得的氧化物层厚度或随后的电容器使用电压。优选的形成电压为1-1000v,特别优选为2-500v,非常特别优选为1-300v。根据本发明制备电容器的方法的第一具体实施方案,形成电压为1-20v,而根据用于制备电容器的方法的第二具体实施方案,形成电压为30-100v。通常使用的多孔电极体的孔隙率优选为10-90%,优选为30-80%,特别优选为50-80%,平均孔径为10-10000nm,优选为20-5000nm,特别优选为50-3000nm。根据本发明方法的具体实施方案,待制备的电解电容器为铝卷绕式电容器。在这种情况下,在工艺步骤a)中,阳极形成多孔铝箔以作为电极材料,形成氧化铝涂层以作为电介质。然后,为以此方式获得的铝箔(阳极箔)提供接触线,并且与同样提供有接触线的另一任选多孔铝箔(阴极箔)一起卷绕,其中这两个箔通过一个或更多隔膜隔开,所述隔膜基于例如纤维素,或者优选基于合成纸。在卷绕后,将以此方式获得的阳极体例如借助胶带固定。可通过在烘箱中加热而将隔膜碳化。用于铝卷绕式电容器的阳极体的该制备方法和方式由现有技术已知,并且例如描述在us7,497,879b2中。根据本发明方法的其他具体实施方案,待制备的电解电容器为铝叠层式电容器或钽电解电容器(“钽elco”),特别是具有聚合物外层的钽电解电容器,例如如de-a-102009007594所述。在本发明方法的工艺步骤ii)中,将可通过本发明第一或第二方法获得的液体组合物、包含具有非常确定的mw/mn值的官能化π共轭聚噻吩的本发明第一液体组合物,或者包含含非常确定的通式(ia)和(ib)重复单元的相对量的官能化π共轭聚噻吩的本发明第二液体组合物,优选pedot-s的水溶液引入阳极体的至少一部分中。在本文中,应当注意的是,在将可通过本发明第一或第二方法获得的液体组合物或本发明的第一或第二液体组合物引入阳极体的至少一部分之前,可将其他组合物,例如pedot/pss分散体引入阳极体中以形成导电层。因此,并非必须要求将可通过本发明第一或第二方法获得的液体组合物或本发明的第一或第二液体组合物直接施加至阳极体的介电层的至少一部分上。将液体组合物通过已知方法引入多孔区域中,例如浸渍、浸涂、倾注、滴流(drippingon)、喷涂、喷雾、刮刀涂覆、刷涂或印刷,例如喷墨印刷、丝网印刷或移印(tamponprinting)。优选地,该引入通过将工艺步骤a)中提供的阳极体浸入液体组合物中并因此用该液体组合物浸渍而进行。浸入液体组合物中或者用液体组合物浸渍优选实施1秒至120分钟,特别优选5秒至60分钟,最优选10秒至15分钟的时间。可例如通过提高或降低压力、振动、超声波或加热来促进液体组合物在阳极体中的引入。除官能化的π共轭聚合物a)、溶剂c)和任选的呈其还原形式的剩余氧化剂b)之外,工艺步骤ii)中使用的液体组合物此外还可包含其他添加剂,例如表面活性剂物质,例如阴离子表面活性剂如烷基苯磺酸和盐、链烷磺酸盐、醇磺酸盐、醚磺酸盐、磺基琥珀酸盐、磷酸酯、烷基醚羧酸或羧酸盐,阳离子表面活性剂如烷基季铵盐,非离子表面活性剂如直链醇乙氧基化物、羰基合成醇乙氧基化物、烷基酚乙氧基化物或烷基聚葡糖苷,特别是可以以商品名和商购获得的表面活性剂;或者助粘剂,例如有机官能硅烷或其水解物,例如3-缩水甘油氧基丙基三烷氧基硅烷、3-氨基丙基三乙氧基硅烷、3-巯基丙基三甲氧基硅烷、3-甲基丙烯酰氧基丙基三甲氧基硅烷、乙烯基三甲氧基硅烷或辛基三乙氧基硅烷;交联剂,例如蜜胺化合物,掩蔽的异氰酸酯,官能硅烷如四乙氧基硅烷,烷氧基硅烷水解物,例如基于四乙氧基硅烷,环氧基硅烷如3-缩水甘油氧基丙基三烷氧基硅烷-聚氨酯,聚丙烯酸酯或聚烯烃分散体。优选地,工艺步骤ii)中使用的液体组合物包含任选提高导电性的其他添加剂,例如含有醚基团的化合物,例如四氢呋喃;含有内酯基团的化合物,例如γ-丁内酯、γ-戊内酯;含有酰胺或内酰胺基团的化合物,例如己内酰胺、n-甲基己内酰胺、n,n-二甲基乙酰胺、n-甲基乙酰胺、n,n-二甲基甲酰胺(dmf)、n-甲基甲酰胺、n-甲基甲酰苯胺、n-甲基吡咯烷酮(nmp)、n-辛基吡咯烷酮、吡咯烷酮;砜和亚砜,例如环丁砜(四亚甲基砜)、二甲亚砜(dmso);糖或糖衍生物,例如蔗糖、葡萄糖、果糖、乳糖;糖醇,例如山梨糖醇、甘露糖醇;呋喃衍生物,例如2-呋喃甲酸、3-呋喃羧酸;甘油、二甘油、三甘油或四甘油。此外,工艺步骤ii)中使用的液体组合物可包含作为添加剂的一种或多种可溶于有机溶剂中的有机粘合剂,如wo2009/141209a1第12页第16-34行所述。用于制备固体电解质层的液体组合物可具有1-14的ph,优选1-8的ph。对于腐蚀敏感的电介质,例如氧化铝或铌氧化物,优选ph为2.5-8的液体组合物,以便不损坏电介质。为了调节ph,可在工艺步骤ii)中使用的液体组合物中添加碱或酸以作为添加剂,如wo2010/003874a2第4页第13-32行所述。这些添加剂不会损害液体组合物的成膜并且在较高温度(例如焊接温度)下不挥发,而是在这些条件下保留在固体电解质中,例如优选碱为2-二甲基氨基乙醇、2,2'-亚氨基二乙醇或2,2',2"-次氮基三乙醇,酸为聚苯乙烯磺酸。取决于施加方法,工艺步骤ii)中使用的液体组合物的粘度可为0.01-1000mpa·s(使用流变仪在20℃和100s-1的剪切速率下测量)。粘度优选为1-500mpa·s,特别优选为1-250mpa·s。在制备铝卷绕式电容器的情况下,粘度非常特别优选为1-200mpa·s,而在钽电解电容器或铝叠层式电容器的制备中,非常特别优选为1-50mpa·s。粘度的调节可例如通过加入适当的流变改性剂作为其他添加剂实现。工艺步骤ii)中使用的液体组合物的固含量优选为0.01-20重量%,特别优选为0.1-15重量%,最优选为0.25-10重量%,在每种情况下基于液体组合物的总重量。液体组合物的固含量通过在足够高的温度下干燥液体组合物以除去溶剂c)而测定。根据本发明的制备电容器的方法的特别优选的实施方案,引入电容器体中的液体组合物不仅包含官能化的π共轭聚合物,而且除了该自掺杂的导电聚合物之外,还包含外掺杂的导电聚合物,优选如wo2014/048562a2中所公开的pedot/pss。就组合使用自掺杂聚合物如pedot-s和外掺杂聚合物如pedot/pss以形成固体电解质而言,在此通过引用将wo2014/048562a2的公开内容并入本文,并且形成本申请公开内容的一部分。在用可通过根据本发明第一或第二方法获得的液体组合物或上文所述的本发明第一或第二液体组合物浸渍阳极体之后,有利的是在随后的工艺步骤iii)中至少部分移除液体组合物中所含的溶剂c),从而形成完全或部分覆盖电介质并因此形成电容器体的固体电解质。在本文中,优选用固体电解质将电介质覆盖至少10%,特别优选至少25%,最优选至少50%,其中覆盖度可通过在120hz下干燥和潮湿状态下的电容器的电容而确定,如de-a-102005043828所述。所述移除或硬化优选通过从液体组合物中取出电极体并将其干燥而进行,干燥优选在20-260℃,特别优选50-220℃,最优选80-200℃的温度下进行。当然也可通过冷冻干燥至少部分移除溶剂c)。工艺步骤ii)和iii)也可重复一次或数次,以便以此方式使沉积在电介质上的固体电解质层的厚度或电极体中的电解质填充程度适应具体要求。在以此方式制备电容器体之后,可通过本领域技术人员已知的方法和方式来进一步修饰电容器体。在钽电解电容器的情况下,如de-a-102004022674或de-a-102009007594所述,电容器体可例如用聚合物外层覆盖,和/或用石墨层和银层覆盖,如de-a-102005043828所知;而在铝卷绕式电容器的情况下,根据us7,497,879b2的教导,将电容器体引入铝烧杯中,提供密封玻璃并通过卷曲机械闭合。然后可以以已知的方式通过老化除去电容器的电介质中的缺陷。对实现上述目的的贡献还可由可通过上述方法做出,优选通过上述方法获得的电容器做出。优选地,该电容器为钽电解电容器或铝电容器,例如铝叠层式电容器或铝卷绕式电容器。对实现上述目的的贡献还由可通过本发明第一或第二方法获得的液体组合物的用途做出,优选通过本发明第一或第二方法获得的液体组合物的用途做出,或者由包含具有非常确定的mw/mn值的官能化π共轭聚噻吩的本发明第一液体组合物的用途做出,或者由包含含非常确定的通式(ia)和(ib)重复单元的相对量的官能化π共轭聚噻吩的本发明第二液体组合物的用途做出,其中它们用于制备电子器件中的导电层,其中电子器件优选选自光导电池、光敏电阻器、光开关、光敏晶体管、光电管、ir检测器、光伏器件、太阳能电池、用于存储器件的涂料材料、场效应电阻器件、抗静电膜、生物传感器、电致变色器件、固体电解质电容器、储能器件、触摸屏和电磁屏蔽。在本文中,特别优选将可通过本发明第一或第二方法获得的液体组合物,优选通过本发明第一或第二方法获得的液体组合物,包含具有非常确定的mw/mn值的官能化π共轭聚噻吩的本发明第一液体组合物,或者包含含非常确定的通式(ia)和(ib)重复单元的相对量的官能化π共轭聚噻吩的本发明第二液体组合物用于制备固体电解质电容器的固体电解质层。在本文中,特别优选将可通过本发明第一或第二方法获得的液体组合物,优选通过本发明第一或第二方法获得的液体组合物,包含具有非常确定的mw/mn值的官能化π共轭聚噻吩的本发明第一液体组合物,或者包含含非常确定的通式(ia)和(ib)重复单元的相对量的官能化π共轭聚噻吩的本发明第二液体组合物用于wo2014/048562a2中所公开的方法中。现在借助于非限制性的附图和实施例更详细地阐述本发明。图1是通过可由本发明的电容器制备方法获得的电容器的一部分的截面图。这具有电极体1,其通常由多孔电极材料2如铝制成。在电极材料2的表面4上,形成作为薄层的电介质3,从而形成仍为多孔的阳极体5,其包含电极材料2的电极体1和形成的电介质3。电介质3之后(任选在其他层之后)为固体电解质6(例如使用本发明方法制备的液体组合物制备的层),由此形成电容器体7,其包括电极材料2的电极体1、电介质3和形成的固体电解质6。测试方法电导率将清洁的玻璃基材铺设在旋涂机上,将10ml的本发明液体组合物分布在基材上。然后通过板的旋转将剩余的溶液旋走。随后,将如此涂布的基材在130℃下在热板上干燥15分钟。然后通过层厚测量设备测定层厚(tencor,alphastep500)。电导率通过以10mm的距离经由阴影掩模气相沉积2.5cm长的ag电极而测定。将用静电计(keithly614)测得的表面电阻乘以层厚,从而获得比电阻率。电导率是比电阻率的倒数。氧含量氧含量用knickportamess911oxy(knickelektronischegmbh&co.kg,beuckestraβe22,德国柏林)测量。在测量之前,将设备相对于环境空气校准。为了测定反应开始时的氧含量,将传感器在氮气流下浸入反应溶液中。氯离子含量氯离子含量使用以下设备和测量条件通过离子色谱法测定:设备:metrohm882compacticplus(metrohmag,ionenstrasse,ch-9100herisau,瑞士)软件:metrohmmagicnet柱:metrosepasupp5;粒度5μm;长度150-25mm;直径5mm洗脱液的制备:将3.2ml1mol/l碳酸钠溶液和1ml碳酸氢钠溶液混合,加入995.8ml去离子水。流速:0.7ml/min温度:室温对于校准,使用以下标样:“chlorid-1.000mg/lcl-inwasser(ausnacl)standardfürdieionenchromatographie”,vwr产品代码458012q100ml(vwrinternationalgmbh,darmstadt)。对于校准,通过用去离子水稀释标样而制备浓度系列,以便在0.1-30ppm的相关范围内校准离子色谱。如果样品的氯离子含量高于30ppm,则用去离子水将样品稀释,直至氯离子浓度达到校准范围。将结果乘以稀释倍数,以计算原始样品的氯离子含量。等效串联电阻(esr)等效串联电阻(mω)借助lcr仪(agilent4284a)在100khz下在20℃下测定。在每个电容器实验中,准备至少5个电容器,并测定平均esr值。电容(cap)电容(μf)借助lcr仪(agilent4284a)在120hz下在20℃下测定。在每个电容器实验中,准备至少5个电容器,并测定平均电容值。mn和mw质均分子量mw和摩尔平均分子量mn通过凝胶渗透色谱法(gpc)使用聚苯乙烯磺酸作为标样测定。与hp1047ari检测器组合地使用具有hp11001312泵和三个后续柱(mcx1.000、mcx100.000和mcx10.000.000)的模块化gpc系统。洗脱液为水。处理温度设定为40℃,使用0.5000ml/min的流动速度。校准标样为聚苯乙烯磺酸。在测量之前,使pedot-s样品通过0.45μm醋酸纤维素注射器过滤器过滤。官能化的π共轭聚噻吩的组成用于制备本发明的包含官能化π共轭聚噻吩的液体组合物的噻吩单体中的通式(ia)和(ib)单体单元的相对量通过hplc测定。d50和d90直径分布的d90值表示官能化π共轭聚噻吩的所有颗粒的总重量的90%可分配给直径小于或等于d90值的那些颗粒。因此,直径分布的d50值表示官能化的π共轭聚噻吩的所有颗粒的总重量的50%可分配给直径小于或等于d50值的颗粒(因此,d50值表示重均粒径)。d50和d90的测定通过超离心机测定进行。平均如果不另外提及,平均值对应于算术平均值。实施例为了制备如下所述的pedot-s溶液,如chevrot等(j.electroanal.chem.1998,443,217-226)所述制备4-(2,3-二氢噻吩并[3,4-b][1,4]二氧杂环己烯-2-基甲氧基)-1-丁磺酸(edot-s)的钠盐并用作单体。合成实施例1(非本发明)由玻璃制成的3l夹套烧杯配有机械搅拌器、温度计和氮气流。组分a在该烧杯中,将243.6g(0.9mol)氯化铁(iii)溶解在800g去离子水中,并在搅拌下将氮气吹入溶液30分钟,直至氧含量低于0.25mg/l。组分b在单独的玻璃烧杯中,将100gedot-s钠盐(0.29mol)溶于1200g去离子水中。通过柔性管将氮气吹入该溶液中直至氧含量低于0.25mg/l。在搅拌下将组分b加入到组分a中。将如此获得的混合物在6小时内加热至90-95℃,并在该温度下再保持15小时。在反应结束后,通过加入去离子水将反应混合物填充至体积为10l,然后通过超滤(pallmicrozaslp1053,截止量为10000g/mol)进行处理,由此移除8l水。重复该过程6次以移除无机盐。由此获得的分散体的特征在于电导率为0.05s/cm,固含量为1.62重量%。合成实施例2(非本发明)由不锈钢制成的3l夹套罐装配有机械搅拌器、位于上盖上的通风阀、可闭合的材料入口和温度计。组分a向该罐中引入2000g去离子水、16.0g10重量%硫酸铁(iii)水溶液和100gedot-s钠盐(0.29mol)。搅拌器以50rpm运行,将温度调节至20℃,将内部压力降至100hpa。随后将罐中的压力升至大气压,随后将压力进一步降低至25hpa以排出氧气。组分b在单独的玻璃烧杯中,将78.5g过二硫酸钠溶解在200ml水中,在搅拌下将氮气吹入溶液30分钟,直至氧含量低于0.25mg/l。然后将组分b吸入罐中。然后关闭材料入口,并借助真空泵将罐的内部压力调节至25hpa。在该减压下继续反应19小时。在反应结束后,通过加入去离子水将反应混合物填充至体积为10l,然后通过超滤(pallmicrozaslp1053,截止量为10000g/mol)进行处理,由此移除8l水。重复该过程6次以移除无机盐。由此获得的分散体的特征在于电导率为0.09s/cm,固含量为1.05重量%。合成实施例3(根据本发明)由不锈钢制成的3l夹套罐装配有机械搅拌器、位于上盖上的通风阀、可闭合的材料入口和温度计。组分a向该罐中引入2000g去离子水、16.0g10重量%的硫酸铁(iii)水溶液,5.7g硫酸(95重量%)和100gedot-s钠盐(0.29mol)。搅拌器以50rpm运行,将温度调节至20℃,将内部压力降至100hpa。随后将罐中的压力升至大气压,随后将压力进一步降低至25hpa以排出氧气。组分b在单独的玻璃烧杯中,将78.5g过二硫酸钠溶解在200ml水中,在搅拌下将氮气吹入溶液30分钟,直至氧含量低于0.25mg/l。然后将组分b吸入罐中。然后关闭材料入口,并借助真空泵将罐的内部压力调节至25hpa。反应溶液的初始ph为1.9,在该减压下继续反应19小时。在反应结束后,通过加入去离子水将反应混合物填充至体积为10l,然后通过超滤(pallmicrozaslp1053,截止量为10000g/mol)进行处理,由此移除8l水。重复该过程6次以移除无机盐。由此获得的组合物的特征在于电导率为27s/cm,固含量为1.22重量%。通过超滤进一步浓缩组合物,直至达到2.4重量%的固含量。合成实施例4(制备pedot/pss分散体;非本发明)首先将868g去离子水和330g平均分子量为70000g/mol,固含量为3.8重量%的聚苯乙烯磺酸水溶液引入具有搅拌器和内部温度计的2l三颈烧瓶中。将反应温度保持在20和25℃之间。在搅拌下加入5.1g3,4-乙撑二氧噻吩。将溶液搅拌30分钟。然后加入0.03g硫酸铁(iii)和9.5g过硫酸钠,将溶液再搅拌24小时。在反应结束后,为了移除无机盐,加入100ml强酸性阳离子交换剂和250ml弱碱性阴离子交换剂,将溶液再搅拌2小时。滤出离子交换剂。将聚(3,4-乙撑二氧噻吩)/聚苯乙烯磺酸盐分散体在700巴的压力下用高压均化器均化10次。随后将分散体浓缩至2.5%的固含量,然后在1500巴的压力下额外再均化5次。合成实施例5(根据现有技术制备pedot-s组合物)在氩气下将0.496gedot-s(1.5mmol)溶于18ml蒸馏水中。然后一次加入0.97g(6.0mmol)fecl3。此后,将溶液在室温下搅拌8小时,并在100℃下加热3小时,冷却并后处理。为了进行后处理,将溶液用蒸馏水稀释至约3重量%,加入9gs100和9gmp62,将混合物在室温下搅拌4小时。在滤出离子交换剂后,得到固含量为2.71%的深蓝色聚合物溶液。对比实施例1(根据wo2014/048562a2制备电容器)将45g合成实施例4的pedot/pss分散体、45g合成实施例5的pedot-s组合物和10g聚乙二醇400(peg-400)混合,用氨将ph调节至3.0(分散体a)。为在36v下形成的具有200mm×5mm尺寸的多孔铝箔(阳极箔)和具有210mm×3mm尺寸的多孔铝箔(阴极箔)各自提供接触线,然后与两张纤维素隔离纸一起卷绕,用胶带固定。制备20个这些氧化的电极体。然后将氧化电极体的隔离纸在300℃的烘箱中碳化。将氧化的电极体在分散体a中浸渍15分钟。此后,在120℃下干燥20分钟,然后在150℃下干燥20分钟。再一次实施浸渍和干燥。测量平均电性能值。实施例1(根据wo2014/048562a2制备电容器)将45g合成实施例4的pedot/pss分散体、45g合成实施例3的pedot-s组合物和10g聚乙二醇400(peg-400)混合,用氨将ph调节至3.0(分散体b)。根据对比实施例1的程序制备电容器。测定平均电性能值,相对于对比实施例1归一化的结果示于表1中。表1capesr对比实施例11.001.00实施例11.010.53合成实施例6(制备用于聚合物外层的pedot/pss分散体)首先将1736g去离子水和660g平均分子量为70000g/mol,固含量为3.8重量%的聚苯乙烯磺酸水溶液引入具有搅拌器和温度计的5l玻璃反应器中。反应温度保持在20和25℃之间。在搅拌下加入10.2g3,4-乙撑二氧噻吩。将溶液搅拌30分钟。然后加入0.06g硫酸铁(iii)和19g过硫酸钠,将溶液再搅拌24小时。在反应结束后,为了移除无机盐,加入200ml强酸性阳离子交换剂和500ml弱碱性阴离子交换剂,将溶液再搅拌2小时。滤出离子交换剂。通过随后浓缩使获得的分散体达到1.5%的固含量。将160g该分散体、28g水、6g磺基聚酯(eastek1100,固含量30%,平均分子量10000-15000,eastman)、8g二甲亚砜、1g3-缩水甘油氧基丙基三甲氧基硅烷(silquesta-187,osispecialties)和0.4g润湿剂(dynol604,airproducts)在玻璃烧杯中用搅拌器剧烈混合1小时。合成实施例7(制备交联剂溶液)将4.0g对甲苯磺酸一水合物、1.7g1,10-二氨基癸烷和95.5g水在玻璃烧杯中用搅拌器剧烈混合。合成实施例8(制备用于钽电解电容器的电极体)在掺入钽丝下,将比电容为18000cv/g的钽粉末压成颗粒并烧结,从而形成尺寸为1.5mm×2.9mm×4.0mm的多孔阳极体。将5个这些多孔阳极体在磷酸电解质中在100v下阳极氧化以形成电介质,从而获得电容器体。合成实施例9通过加入去离子水将合成实施例5的组合物稀释至2.0%的浓度。合成实施例10通过加入去离子水将合成实施例3的组合物稀释至2.0%的浓度。对比实施例2将合成实施例8的电容器体在合成实施例9的组合物中浸渍1分钟。此后,在120℃下干燥10分钟。再实施浸渍和干燥9次。然后将电容器体浸渍在合成实施例7的溶液中。然后,在120℃下干燥10分钟。然后将电容器体浸渍在合成实施例6的分散体中。然后在120℃下干燥10分钟。然后将电容器体浸渍在合成实施例7的溶液中。然后在120℃下干燥10分钟。然后将电容器体浸渍在合成实施例6的分散体中。然后在120℃下干燥10分钟。然后将电容器体浸渍在合成实施例7的溶液中。然后在120℃下干燥10分钟。然后将电容器体浸渍在合成实施例6的分散体中。然后在120℃下干燥10分钟。然后用石墨层覆盖电容器体,然后用银层覆盖,从而以此方式获得成品电容器。确定电性能参数(cap,esr)的平均值。实施例2如对比实施例2所述对电容器体进行处理,然而使用合成实施例10的组合物代替合成实施例9的组合物。确定电性能参数(cap,esr)的平均值,相对于对比实施例2归一化的结果示于表2中。表2capesr对比实施例21.001.00实施例21.150.25合成实施例11(非本发明)在氩气下将0.496gedot-s(1.5mmol)溶于18ml蒸馏水中。然后一次加入0.97g(6.0mmol)fecl3。此后,将溶液在室温下搅拌8小时,并在100℃下加热3小时,冷却并后处理。为了进行后处理,将溶液用蒸馏水稀释至1重量%,加入9gs100和9gmp62,将混合物在室温下搅拌4小时。在滤出离子交换剂后,得到固含量为1重量%的深蓝色聚合物溶液。在由此获得的组合物中,通过蒸发将固含量调节至2.15重量%。合成实施例12(根据本发明)由不锈钢制成的3l夹套罐装配有机械搅拌器、位于上盖上的通风阀、可闭合的材料入口和温度计。组分a向该罐中引入2000g去离子水、8.0g10重量%硫酸铁(iii)水溶液、2.9g硫酸(95重量%)和50gedot-s钠盐(0.15mol)。搅拌器以50rpm运行,将温度调节至20℃,将内部压力降至100hpa。随后将罐中的压力升至大气压,随后将压力进一步降低至25hpa以排出氧气。组分b在单独的玻璃烧杯中,将39.3g过二硫酸钠溶解在200ml水中,并在搅拌下将氮气吹入溶液30分钟,直至氧含量低于0.25mg/l。然后将组分b吸入罐中。然后关闭材料入口,并借助真空泵将罐的内部压力调节至25hpa。反应溶液的初始ph为1.9,在该减压下继续反应19小时。在反应结束后,加入600glewatitmonopluss108h和500glewatitmp62(lanxessag,cologne),并用机械搅拌器搅拌。在6小时后,通过过滤移除lewatit。借助旋转蒸发器浓缩样品,直至达到>2重量%的固含量。由此获得的组合物的特征在于固含量为2.14重量%。合成实施例13(根据本发明)由不锈钢制成的3l夹套罐装配有机械搅拌器、位于上盖上的通风阀、可闭合的材料入口和温度计。组分a向该罐中引入2000g去离子水、16.0g10重量%的硫酸铁(iii)水溶液、5.7g硫酸(95重量%)和100gedot-s钠盐(0.29mol)。搅拌器以50rpm运行,将温度调节至20℃,将内部压力降至100hpa。随后将罐中的压力升至大气压,随后将压力进一步降低至25hpa以排出氧气。组分b在单独的玻璃烧杯中,将78.5g过二硫酸钠溶解在200ml水中,在搅拌下将氮气吹入溶液30分钟,直至氧含量低于0.25mg/l。然后将组分b吸入罐中。然后关闭材料入口,并借助真空泵将罐的内部压力调节至25hpa。反应溶液的初始ph为1.9,在该减压下继续反应19小时。在反应结束后,加入1100glewatitmonopluss108h和1000glewatitmp62(lanxessag,cologne)并用机械搅拌器搅拌。在6小时后,通过过滤移除lewatit。由此获得的组合物的特征在于固含量为1.19重量%。借助旋转蒸发器进一步浓缩组合物,直至达到>2重量%的固含量。由此获得的组合物的特征在于固含量为2.15%,电导率为41s/cm。在合成实施例11、合成实施例12和合成实施例13中得到的组合物的质均分子量mw和摩尔平均分子量mn示于下表3中:表3mwmnmw/mn合成实施例1123300g/mol5300g/mol4合成实施例1279000g/mol8400g/mol9合成实施例13204000g/mol9500g/mol21对比实施例3如对比实施例1那样制备铝电容器,唯一的不同之处在于使用合成实施例11的组合物代替合成实施例5的组合物。实施例3如实施例1那样制备铝电容器,唯一的不同之处在于使用合成实施例12的组合物代替合成实施例3的组合物。实施例4如实施例1那样制备铝电容器,唯一的不同在于使用合成实施例13的组合物代替合成实施例3的组合物。电性能参数(cap,esr)的平均值示于表4中,其中的值相对于对比实施例3归一化。还显示了电容器在85℃和85%相对湿度下储存500小时后的esr值(所述值相对于在这些条件下储存之前的相应值归一化)。表4这些结果清楚地表明,当使用具有大于6的高mw/mn值的pedot-s组合物来制备电容器中的固体电解质层时,不仅可以提高esr值(这可从表4第3列看出),而且可以提高电容器在高温和高相对湿度下储存时的稳定性。从表4第4列可以看出,与固体电解质层使用现有技术的pedot-s组合物制备的电容器相比,固体电解质层用本发明的pedot-s组合物制备(即,使用具有mw/mn值>6的pedot-s组合物)的电容器的esr值提高程度较低。合成实施例14(制备用于钽电解电容器的电极体)在引入钽丝下,将比电容为30000cv/g的钽粉末压成颗粒并烧结,从而形成尺寸为1.4mm×2.8mm×3.9mm的多孔阳极体。将5个这些多孔阳极体在磷酸电解质中在60v下阳极氧化以形成电介质,从而获得电容器体。对比实施例4如对比实施例2那样制备钽电解电容器,不同之处在于,使用合成实施例14的电容器体代替合成实施例8的电容器体,使用合成实施例11的组合物代替合成实施例9的组合物。实施例5如实施例2那样制作钽电解电容器,不同之处在于,使用合成实施例14的电容器体代替合成实施例8的电容器体,使用合成实施例12的组合物代替合成实施例10的组合物。实施例6如实施例2那样制备钽电解电容器,不同之处在于,使用合成实施例14的电容器体代替合成实施例8的电容器体,使用合成实施例13的组合物代替合成实施例10的组合物。电性能参数(cap,esr)的平均值示于表5中,其中的值相对于对比实施例4归一化。表5capesr对比实施例41.001.00实施例51.000.57实施例61.170.26这些结果清楚地表明,当使用具有大于6的高mw/mn值的pedot-s组合物来制备电容器中的固体电解质层时,可以提高电容和esr值。合成实施例15(非本发明)由不锈钢制成的3l夹套罐装配有机械搅拌器、位于上盖上的通风阀、可闭合的材料入口和温度计。组分a向该罐中引入2000g去离子水、16.0g10重量%的硫酸铁(iii)水溶液、5.7g硫酸(95重量%)和100g含有20重量%prodot-s钠盐(通过hplc测定)的edot-s钠盐(0.29mol)。搅拌器以50rpm运行,将温度调节至20℃,将内部压力降至100hpa。随后将罐中的压力升至大气压,随后将压力进一步降低至25hpa以排出氧气。组分b在单独的玻璃烧杯中,将78.5g过二硫酸钠溶解在200ml水中,在搅拌下将氮气吹入溶液30分钟,直至氧含量低于0.25mg/l。然后将组分b吸入罐中。然后关闭材料入口,并借助真空泵将罐的内部压力调节至25hpa。反应溶液的初始ph为1.9,在该减压下继续反应19小时。在反应结束后,通过加入去离子水将反应混合物填充至体积为10l,然后通过超滤(pallmicrozaslp1053,截止量10000g/mol)进行处理,由此移除8l水。重复该过程6次以移除无机盐。所得分散体的固含量为1.47重量%,借助旋转蒸发器进一步浓缩至固含量为2.96重量%。合成实施例16(根据本发明)以与合成实施例15相同的方式制备pedot-s组合物,唯一的不同之处在于使用含有10重量%prodot-s钠盐的edot-s钠盐。对比实施例5如对比实施例4那样制备钽电解电容器,唯一的不同之处在于,使用合成实施例15的组合物代替合成实施例11的组合物。实施例7如实施例5中制备的钽电解电容器,唯一的不同之处在于,使用合成实施例16的组合物代替合成实施例10的组合物。电性能参数(cap,esr)的平均值示于表6中,其中的值相对于对比实施例5归一化。表6capesr对比实施例51.001.00实施例71.000.77这些结果清楚地表明,与固体电解质通过基于edot-s-单体具有20重量%prodot-s含量的pedot-s组合物制备的钽电解电容器相比,当使用具有仅10重量%prodot-s含量的pedot-s组合物来制备钽电解电容器中的固体电解质层时,esr值显著更低。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种具有阻燃功能的复合纱线的制作方法
- 一种具有阻燃功能的面料的制作方法
- 制动蹄自动铆压设备的密封橡胶圈的制作方法
- 一种混合脂肪基双(苯并咪唑啉季铵盐)及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗缺氧药物的制作方法
- 二氮芴基芳香型二酸单体及其聚苯并咪唑聚合物的合成的制作方法
- 一种苯并三唑基?亚烷基双酚化合物的制备方法