一种5-甲基-3,4-二苯基异噁唑的合成方法与流程
[0001]
本发明属于药物技术领域,特别涉及多种新型抗癌药物中间体5-甲基-3,4-二苯基异噁唑的合成。
技术背景
[0002]
5-甲基-3,4-二苯基异噁唑,cas:37928-17-9,其结构式如下:
[0003][0004]
异噁唑类化合物具有非常良好的生物活性,在农业、医药领域材料化学应用上具有广阔的应用价值,其显示如抗菌、抗炎、降血压等药物活性作用,而3,4
--
二芳基异噁唑作为药物骨架被广泛应用于生物制药,例如蛋白激酶抑制剂、非甾体抗炎药物等。其中5-甲基-3,4-二苯基异噁唑是镇痛药物帕瑞昔布钠重要中间体,得益于其镇痛应用的广泛,副作用小,甚至可以代替或减少吗啡的用量,其需求量日益增长。
[0005]
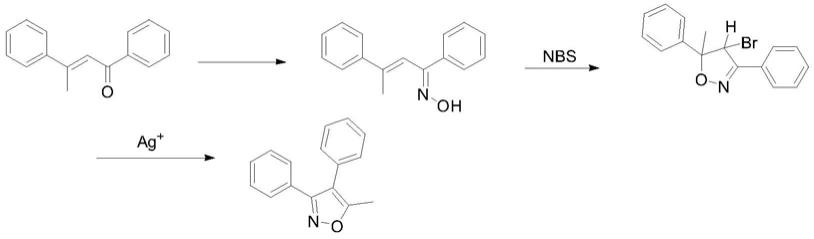
其中[journal ofheterocyclic chemistry,1980,vol.17,p.475-479]原料经肟化、环化及重排消除得到产品,但其原料成本很高及用到银催化剂进行重排,路线如下:
[0006][0007]
上述合成方法中,原料不易获得,或者采用铟催化剂合成原料。因此,采用易得的起始原料,对合成工艺进行优化,寻找到合适的放大工艺,提高产品的市场竞争力是非常必要的。
技术实现要素:
[0008]
本发明针对上述问题,开发了一种5-甲基-3,4-二苯基异噁唑新合成路线。以二苯甲酰甲烷为原料与羟胺生成1,3-二苯基丙烷-1,3-二酮单肟,随后与甲基格氏试剂对羰基进行加成,接着环氧化得到(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟,最后在lewis催化环化、脱水重排得到5-甲基-3,4-二苯基异噁唑。采用本发明工艺路线,起始原料易得,工艺过程中均为精细化工中常见单元操作,反应连续性增加,方便了工业化的操作,为下游药物的大规模应用提供基础。
[0009]
本发明所述一种5-甲基-3,4-二苯基异噁唑的合成方法,包括如下步骤:以二苯甲酰甲烷1为原料,经过肟化得到1,3-二苯基丙烷-1,3-二酮单肟2,随后与甲基格氏试剂对羰
基进行加成,接着环氧化得到(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟4,最后在lewis催化作用下环化、脱水重排得到5-甲基-3,4-二苯基异噁唑。采用反应方程式表示如下:
[0010][0011]
本发明所述合成方法,从5-甲基-3,4-二苯基异噁唑依次经过肟化、加成环氧化、重排脱水等三步完成,具体反应步骤为:
[0012]
第一步:肟化反应,将二苯甲酰甲烷1溶于溶液中,与羟胺生成1,3-二苯基丙烷-1,3-二酮单肟2。
[0013]
进一步地,在上述技术方案中,第一步中,羟胺为硫酸羟胺或盐酸羟胺,有机溶液采用甲醇/水或乙醇/水。反应温度为25-32℃。
[0014]
进一步地,在上述技术方案中,第一步中,二苯甲酰甲烷、羟胺与催化剂的摩尔比为1:0.95-1:0.3-0.5。
[0015]
第二步:将中间体1,3-二苯基丙烷-1,3-二酮单肟溶于有机溶剂中,-15~0℃滴加甲基格氏试剂,淬灭后有机相加入三氟乙酸或对甲苯磺酸脱水,随后有机相加入环氧化试剂,得到中间体4(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟。
[0016]
进一步地,在上述技术方案中,甲基格氏试剂选自甲基氯化镁或甲基溴化镁,溶剂采用四氢呋喃或甲基四氢呋喃,消除用酸为三氟乙酸或对甲苯磺酸,环氧化试剂为间氯过氧苯甲酸或叔丁基过氧化氢。
[0017]
进一步地,在上述技术方案中,第二步1,3-二苯基丙烷-1,3-二酮单肟、甲基格氏试剂、环氧化试剂摩尔比为1:2.5-3.0:1.05-1.2。
[0018]
第三步:中间体4(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟溶于二氯甲烷中,滴加lewis酸,反应淬灭后有机相加入三氟乙酸或对甲苯磺酸,温度控制在35-40℃下反应2-5小时得到5-甲基-3,4-二苯基异噁唑。
[0019]
进一步地,在上述技术方案中,lewis酸选自三氟化硼乙醚络合物或三氯化硼。
[0020]
进一步地,在上述技术方案中,(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟、lewis酸摩尔比为1:4.0-5.0。
[0021]
发明有益效果:
[0022]
采用本发明工艺路线,每步反应可以连续进行,仅需在最后一步进行重结晶纯化,三步收率在62-65%,最终产品纯度在99.0%以上。
[0023]
路线中起始原料易得,工艺过程中均为精细化工中常见单元操作,反应连续性增加,方便了工业化的操作,为下游药物的大规模应用提供基础。
具体实施例:
[0024]
实施例1
[0025]
第一步:1,3-二苯基丙烷-1,3-二酮单肟的合成
[0026][0027]
氮气保护下,将原料ⅰ112g(0.5mol,1eq)加入到25%甲醇水溶液1000ml中,控制温度25-32℃,分批加入盐酸羟铵68.8g(0.495mol,0.99),并通过滴加醋酸钠水溶液调节ph=5.7,添加完毕后,在25℃下反应24小时,静置,取上清液原料≤2%,35-45℃减压浓缩,蒸除甲醇,降温至20-25℃,过滤,滤饼用水淋洗,35-45℃真空干燥,得到1,3-二苯基丙烷-1,3-二酮单肟112g,hplc检测化学纯度96.1%,收率93.6%,1hnmr(400mhz,dmso-d6):δ=7.86(m,2h),7.67-7.45(m,3h),7.37-7.30(m,5h),6.43(s,1h),3.98(m,2h).
[0028][0029]
氮气保护下,将原料ⅰ112g(0.5mol,1eq)加入到32%乙醇水溶液1000ml中,控制温度25-32℃,分批加入硫酸羟铵64.3g(0.49mol,0.98),并通过滴加二乙胺调节ph=5.3-5.8,添加完毕后,在25℃下反应24小时,静置,取上清液原料≤2%,35-45℃减压浓缩,蒸除乙醇,降温至20-25℃,过滤,滤饼用水淋洗,35-45℃真空干燥,得到1,3-二苯基丙烷-1,3-二酮单肟110.7g,hplc检测化学纯度97.9%,收率92.5%。
[0030]
实施例2
[0031]
第二步:(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟的合成。
[0032][0033]
氮气保护下,将中间体ⅱ112g溶于500g四氢呋喃中,降温至-15℃,滴加(2m/l)甲基氯化镁四氢呋喃溶液665.2ml,滴加结束后,缓慢升温至5℃,反应1小时,加入饱和氯化铵水溶液进行淬灭,静置分层,有机相用二氯甲烷500ml萃取,有机相合并,加入三氟乙酸102g,升温至45℃反应5h后,取样lc中间体4≥87%,减压浓缩至剩余1体积,降温至10-25℃,加入正庚烷300g打浆,过滤,烘干得到中间体(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟100.4g,hplc检测化学纯度97.3%。收率85.2%,1hnmr(400mhz,dmso-d6):δ=7.60-7.19(m,10h),6.32(s,1h),2.9(s,1h),1.59(s,3h).
[0034][0035]
氮气保护下,将中间体2112g溶于500g四氢呋喃中,降温至-15℃,滴加(2m/l)甲基溴化镁四氢呋喃溶液665.2ml,滴加结束后,缓慢升温至5℃,反应1小时,加入饱和氯化铵水溶液进行淬灭,静置分层,有机相用二氯甲烷500ml萃取,有机相合并,加入对甲苯磺酸一水合物133g,升温至35℃反应2h后,取样lc中间体4≥87%,加入1%盐酸水溶液300ml*3水洗,有机相减压浓缩至剩余1体积,降温至10-25℃,加入正庚烷300g打浆,过滤,烘干得到中间体(3-甲基-3-苯基-环氧乙烷)-苯基甲酮肟102.1g,hplc检测化学纯度94.2%。收率86.7%,
[0036]
实施例3
[0037]
第三步:5-甲基-3,4-二苯基异噁唑的合成。
[0038][0039]
氮气保护下,将中间体4100g(0.395mol,1eq)溶于700ml二氯甲烷中,降温至0℃,滴加46%三氟化硼乙醚络合物511.9g(1.659mol,4.2eq),缓慢升温至20℃,反应2h。取样lc检测原料<1%,反应液降温至0-10℃,将反应液加入到1n盐酸500ml水淬灭,静置分层,水相用二氯甲烷500ml萃取,合并有机相体,有机相减压浓缩至剩余3体积,再加入三氟乙酸109g,升温至40-45℃反应6小时,取样lc检测中间体5≤1%,减压浓缩至剩余2体积,加入正庚烷析出,过滤得到粗品。粗品经乙醇水重结晶得到5-甲基-3,4-二苯基异噁唑84g,收率90.4%,mass(e+1)=236.11。
[0040]1hnmr(400mhz,dmso-d6):δ=7.48(m,4h),7.32(m,4h),7.22(m,2h),2.35(s,3h).
[0041][0042]
氮气保护下,将中间体4100g(0.395mol,1eq)溶于700ml二氯甲烷中,降温至-10℃,通入三氯化硼129.6g(1.106mol,2.8eq),缓慢升温至20℃,反应1h。取样lc检测原料<1%,反应液降温至0-10℃,将反应液加入到1000ml水淬灭,静置分层,水相用二氯甲烷500ml萃取,合并有机相体,有机相减压浓缩至剩余3体积,再加入对甲苯磺酸一水合物123g,升温至40-45℃反应6小时,取样lc检测中间体5≤1%,加入1%盐酸水溶液300ml水洗,有机相减压浓缩至剩余2体积,加入正庚烷析出,过滤得到粗品。粗品经乙醇水重结晶得到5-甲基-3,4-二苯基异噁唑79.1g,收率85.1%,mass(e+1)=236.11。
[0043]
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原
理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1