结晶型硝羟喹啉及其制备方法和用途与流程
1.本发明涉及结晶型硝羟喹啉及其制备方法和用途。
背景技术:
2.硝羟喹啉(nitroxoline),化学名为5-硝基-8-羟基喹啉,在六十年代曾被开发为口服抗生素药物,主要用于泌尿系统感染,具有较安全的使用历史,后来由于新型抗生素的发现和使用而被取代。最近几年新的研究发现,硝羟喹啉可同时抑制血管内皮细胞中的甲硫氨酸氨基肽酶metap2和沉默信息调节因子2相关酶类sirt1,发挥肿瘤血管新生的协同抑制效应,同时还对肿瘤细胞的增殖有抑制作用。因此,硝羟喹啉又重新被开发用于治疗包括膀胱癌在内的肿瘤。
3.药物的多晶型已经成为药物研发和药品质量控制中必不可少的重要组成部分。对药物多晶型的研究,有助于药物化合物生物活性的选择,有助于增加药物稳定性、溶解性等性质,进而有利于药物制剂的开发以及药品的储存,提高药品生产质量等,还可提高化合物的生物利用度,增进临床疗效。
4.目前,尚未有关于硝羟喹啉晶型的研究和报道。
技术实现要素:
5.本发明所要解决的技术问题是提供一种结晶型硝羟喹啉及其制备方法和用途。本发明人经过了大量的实验研究才发现了能够制得硝羟喹啉结晶的制备方法,并对所得结晶进行了x射线粉末衍射和dvs检测,结果意外发现了无吸湿性的且稳定性良好的硝羟喹啉结晶。
6.本发明通过以下技术方案解决上述技术问题:
7.本发明提供一种硝羟喹啉的a型结晶,其x射线粉末衍射图谱包括位于11.3
°±
0.2
°
、12.9
°±
0.2
°
、16.9
°±
0.2
°
、19.7
°±
0.2
°
、22.1
°±
0.2
°
、23.7
°±
0.2
°
、24.2
°±
0.2
°
、25.9
°±
0.2
°
、27.1
°±
0.2
°
和28.0
°±
0.2
°
的2θ衍射角处的特征峰。
8.本发明还提供一种硝羟喹啉的a型结晶,其x射线粉末衍射图谱包括位于11.3
°±
0.2
°
、12.9
°±
0.2
°
、16.9
°±
0.2
°
、18.1
°±
0.2
°
、19.7
°±
0.2
°
、21.2
°±
0.2
°
、22.1
°±
0.2
°
、22.8
°±
0.2
°
、23.7
°±
0.2
°
、24.2
°±
0.2
°
、25.9
°±
0.2
°
、27.1
°±
0.2
°
、28.0
°±
0.2
°
、29.4
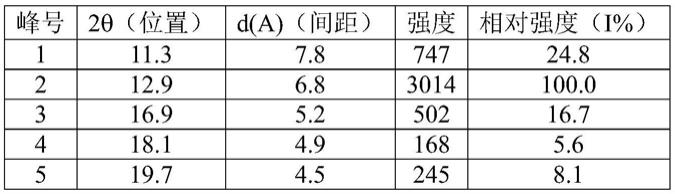
°±
0.2
°
和29.9
°±
0.2
°
的2θ衍射角处的特征峰。
9.本发明还提供一种硝羟喹啉的a型结晶,其x射线粉末衍射图谱如图1所示。
10.本发明还提供一种前述的硝羟喹啉的a型结晶的制备方法(也称为反溶剂法),其包括如下步骤:含有硝羟喹啉和有机溶剂的溶液i与反溶剂接触,析晶,固液分离,即得。
11.在一些优选的实施方案中,所述溶液i由硝羟喹啉和有机溶剂组成。
12.在一些优选的实施方案中,所述有机溶剂选自二甲亚砜、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、c1-c4脂肪酸、c3-c6脂肪酮和c1-c4脂肪醇中的一种或多种,优选为二甲亚砜、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、乙酸、丙酮和甲醇中的一种或多种,更优选为二甲亚
砜、n,n-二甲基甲酰胺和n-甲基吡咯烷酮中的一种或多种,进一步更优选为二甲亚砜。
13.在一些优选的实施方案中,所述硝羟喹啉与所述有机溶剂的质量体积比为5:1至100:1,例如50:0.6,单位为mg/ml。
14.在一些优选的实施方案中,所述反溶剂为水。
15.在一些优选的实施方案中,所述有机溶剂与所述反溶剂的体积比为1:6至1:2,例如1:3、1:4或1:5。
16.在一些优选的实施方案中,所述固液分离为离心分离。所述离心分离的转速优选为3500-4500r/min,例如4000r/min。所述离心分离的时间优选为4-6min,例如5min。
17.在一些优选的实施方案中,所述固液分离之后还包括干燥。所述干燥的温度优选为30-50℃,例如40℃。
18.在一些优选的实施方案中,所述有机溶剂为二甲亚砜,所述反溶剂为水,且二甲亚砜与水的体积比为1:4;或者,
19.所述有机溶剂为n,n-二甲基甲酰胺,所述反溶剂为水,且n,n-二甲基甲酰胺与水的体积比为1:6;或者,
20.所述有机溶剂为n-甲基吡咯烷酮,所述反溶剂为水,且n-甲基吡咯烷酮与水的体积比为1:3;或者,
21.所述有机溶剂为乙酸,所述反溶剂为水,且乙酸与水的体积比为1:2;或者,
22.所述有机溶剂为丙酮,所述反溶剂为水,且丙酮与水的体积比为1:5;或者,
23.所述有机溶剂为甲醇,所述反溶剂为水,且甲醇与水的体积比为1:5。
24.本发明还提供一种前述的硝羟喹啉的a型结晶的制备方法(也称为反溶剂法),其包括如下步骤:
25.(1)于室温,将硝羟喹啉溶解于有机溶剂中,得到澄清溶液;
26.(2)向步骤(1)得到的澄清溶液中逐渐加入反溶剂,析晶,得到晶体;
27.(3)将步骤(2)得到的晶体干燥,得到硝羟喹啉的a型结晶。
28.在一些优选的实施方案中,步骤(1)中,所述有机溶剂选自二甲亚砜、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、c1-c4脂肪酸、c3-c6脂肪酮和c1-c4脂肪醇中的一种或多种,优选为二甲亚砜、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、乙酸、丙酮和甲醇中的一种或多种,更优选为二甲亚砜、n,n-二甲基甲酰胺和n-甲基吡咯烷酮中的一种或多种,进一步更优选为二甲亚砜。
29.在一些优选的实施方案中,步骤(1)中,所述硝羟喹啉与所述有机溶剂的质量体积比为5:1至100:1,例如50:0.6,单位为mg/ml。
30.在一些优选的实施方案中,步骤(2)中,所述反溶剂为水。
31.在一些优选的实施方案中,步骤(1)中的有机溶剂与步骤(2)中的反溶剂的体积比为1:6至1:2,例如1:4。
32.在一些优选的实施方案中,步骤(2)中,所述析晶之后进行固液分离得所述晶体。所述固液分离优选为离心分离。所述离心分离的转速优选为3500-4500r/min,例如4000r/min。所述离心分离的时间优选为4-6min,例如5min。
33.在一些优选的实施方案中,步骤(3)中,所述干燥的温度为30-50℃,例如40℃。
34.在一些优选的实施方案中,步骤(1)中的有机溶剂为二甲亚砜,步骤(2)中的反溶
剂为水,且二甲亚砜与水的体积比为1:4;或者,
35.步骤(1)中的有机溶剂为n,n-二甲基甲酰胺,步骤(2)中的反溶剂为水,且n,n-二甲基甲酰胺与水的体积比为1:6;或者,
36.步骤(1)中的有机溶剂为n-甲基吡咯烷酮,步骤(2)中的反溶剂为水,且n-甲基吡咯烷酮与水的体积比为1:3;或者,
37.步骤(1)中的有机溶剂为乙酸,步骤(2)中的反溶剂为水,且乙酸与水的体积比为1:2;或者,
38.步骤(1)中的有机溶剂为丙酮,步骤(2)中的反溶剂为水,且丙酮与水的体积比为1:5;或者,
39.步骤(1)中的有机溶剂为甲醇,步骤(2)中的反溶剂为水,且甲醇与水的体积比为1:5。
40.特别地,通过反溶剂法制备的硝羟喹啉的a型结晶,收率更高,粒度更细小。
41.本发明还提供一种前述的硝羟喹啉的a型结晶的方法(也称为降温析晶法),其包括如下步骤:将含硝羟喹啉和有机溶剂的溶液ii降温,在混合下析晶,固液分离,即得。
42.在一些优选的实施方案中,所述溶液ii由硝羟喹啉和有机溶剂组成。
43.在一些优选的实施方案中,所述有机溶剂选自甲醇、乙腈、异丙醇、乙酸乙酯、丙酮、甲基叔丁基醚和甲苯中的一种或多种。
44.在一些优选的实施方案中,所述硝羟喹啉与所述有机溶剂的质量体积比为10:3至25:1,单位为mg/ml。
45.在一些优选的实施方案中,所述降温的终点温度为室温至-20℃。
46.在一些优选的实施方案中,所述混合为搅拌。
47.在一些优选的实施方案中,所述混合的时间为0.4-0.6h,例如0.5h。
48.在一些优选的实施方案中,所述固液分离为离心分离。所述离心分离的转速优选为3500-4500r/min,例如4000r/min。所述离心分离的时间优选为4-6min,例如5min。
49.在一些优选的实施方案中,所述固液分离之后还包括干燥。所述干燥的温度优选为30-50℃,例如40℃。
50.本发明还提供一种前述的硝羟喹啉的a型结晶的方法(也称为降温析晶法),其包括如下步骤:
51.(1)在加热下,优选在50-60℃,将硝羟喹啉溶解于有机溶剂中,得到澄清溶液;
52.(2)将步骤(1)得到的澄清溶液降温,优选将至-20℃至室温,并在搅拌下析晶,得到晶体;
53.(3)将步骤(2)得到的晶体干燥,得到硝羟喹啉的a型结晶。
54.在一些优选的实施方案中,根据本发明所述的硝羟喹啉的a型结晶的制备方法,其中步骤(1)中的有机溶剂选自c1-c4脂肪醇、乙腈、丙酮、乙酸乙酯、甲基叔丁基醚和甲苯中的一种或多种,优选c1-c4脂肪醇。
55.在一些优选的实施方案中,步骤(1),硝羟喹啉与有机溶剂的质量体积比为1:40-300,单位为g/ml。
56.本发明还提供一种药物组合物,其包含前述的硝羟喹啉的a型结晶以及药学上可接受的载体。
57.本发明还提供前述的硝羟喹啉的a型结晶或前述的药物组合物在制备治疗癌症的药物中的用途。
58.上述用途中,所述癌症例如为膀胱癌。
59.本文所述的“常温”指的是10-30℃。
60.本文所述的“c1-c4脂肪酸”是指一端含有羧基的脂肪族碳氢链,其具有1-4个碳原子。c1-c4脂肪酸的例子包括但不限于甲酸、乙酸、丙酸和丁酸。
61.本文所述的“c3-c6脂肪酮”是指羰基与两个脂肪烃基相连而成的化合物,其具有3-6个碳原子。c3-c6脂肪酮的例子包括但不限于丙酮、丁酮、戊酮和己酮。
62.本文所述的“c1-c4脂肪醇”是指羟基与脂肪烃基相连而成的化合物,其具有1-4个碳原子。c1-c4脂肪醇的例子包括但不限于甲醇、乙醇、丙醇和丁醇。
63.术语“药学上可接受的”指的是其用于制备药物组合物,所述药物组合物一般是安全的,无毒的,在生物学方面满足需要并且所述药物组合物可以被接受用于兽类或人类药物用途。
64.本文所述的“载体”是指与化合物一起施用的稀释剂、佐剂或赋形剂。药学上可接受的载体可以是液体,例如水和油,包括石油、动物、植物或合成来源的油,例如花生油、大豆油、矿物油、菜籽油。药学上可接受的载体也可以是生理盐水、阿拉伯树胶、明胶、淀粉糊、滑石粉、角蛋白、硅胶、尿素。另外,还可以使用辅助剂、稳定剂、增稠剂、润滑剂和着色剂。
65.本领域技术人员能够理解,本发明的药物组合物可以根据具体的施用方式被制成各种本领域熟知的制剂形式,例如口服剂型(粉剂、片剂、胶囊、软胶囊、液体药物、糖浆、酏丸、散剂、囊剂、粒剂),或局部施用制剂(乳膏、软膏、洗剂、凝胶、香脂、膏药、糊剂、喷雾剂、气雾剂等等),或注射制剂(溶液、悬浮剂、乳剂)。本发明的药物组合物,尤其可以提及的是适合于口服、胃肠外(静脉内或皮下)或鼻部给药的那些剂型,例如,片剂或糖衣丸、舌下片、明胶胶囊、锭剂、栓剂、霜剂、软膏剂、皮肤凝胶、可注射制剂、可饮用的混悬液。
66.本发明的药物组合物可以包含药学上可接受的载体、佐剂或稀释剂,例如填充剂、崩解剂、润滑剂、助悬剂、粘合剂、甜味剂、矫味剂、防腐剂、基质。其中,填充剂例如可为淀粉、预胶化淀粉、乳糖、甘露醇、甲壳素、微晶纤维素、蔗糖;崩解剂例如可为淀粉、预胶化淀粉、微晶纤维素、羧甲基淀粉钠、交联聚乙烯吡咯、低取代羟丙纤维素、交联羧甲基纤维素钠;润滑剂例如可为硬脂酸镁、十二烷基硫酸钠、滑石粉、二氧化硅;助悬剂例如可为聚乙烯吡咯烷酮、微晶纤维素、蔗糖、琼脂、羟丙基甲基纤维素;粘合剂例如可为淀粉浆、聚乙烯吡咯烷酮、羟丙基甲基纤维素。本发明的组合物可以通过利用本领域任何已知方法制成,以使病人用药后能提供快速、持久或缓慢释放的活性成分。
67.本发明的药物组合物通过各种途径给药至个体动物如哺乳动物(大鼠、小鼠、驯化动物或人类),所有的给药方式均是常规的,例如,可以是口服给药、局部给药、直肠给药或者经静脉、肌肉内、经皮、鞘膜内、硬膜外或脑室内注射给药。
68.本发明活性成分的给药剂量可以根据个体的情况和重量、病情的性质和严重程度、药物形式、给药途径以及给药周期的不同而不同,其也可以由本领域技术人员根据动物试验或临床试验选择。本发明活性成分的给药剂量可以在1-100mg/天之间改变,也可在10-1000mg/天之间改变,可以每天单次给药或每天分多次给药。
69.本发明的硝羟喹啉的a型结晶,无吸湿性,具有良好的稳定性,进而有助于给药途
径的选择,以及药物制剂工艺的优化,从而提高药品生产质量,提高药品作用效果。
附图说明
70.图1为本发明硝羟喹啉a型结晶的x射线粉末衍射(xrpd)图谱;
71.图2为本发明硝羟喹啉a型结晶的热重分析(tga)图谱;
72.图3为本发明硝羟喹啉a型结晶的差示扫描量热法(dsc)图谱;
73.图4为本发明硝羟喹啉a型结晶的动态水分吸附(dvs)图谱。
具体实施方式
74.以下将结合实施例更详细地阐述本发明,本发明的实施例仅用于说明本发明的技术方案,并非限定本发明的实质和范围。
75.一、实验仪器
76.1、x-射线粉末衍射(xrpd)
77.仪器型号:shimadzu xrd-6000
78.射线:cu-kα靶
79.电压:40kv,电流:30ma
80.扫描速度:5
°
/min
81.扫描范围:5
°-
50
°
(2θ)
82.2、热重分析(tga)
83.仪器型号:perkinelmer pyris 1tga
84.吹扫气:氮气
85.升温速率:20℃/min
86.温度范围:30℃-250℃
87.3、差示扫描量热仪(dsc)
88.仪器型号:mettler toledodsc3
89.吹扫气:氮气
90.升温速率:20℃/min
91.温度范围:30℃-300℃
92.4、动态水分吸附(dvs)
93.仪器型号:sms dvs intrinsic
94.测试温度:25℃
95.平衡时间:dm/dt:0.01%/min;
96.相对湿度变化范围:0%-95%-0%
97.rh(%)测试每步湿度变化:5%
98.二、实验试剂:
99.硝羟喹啉由起始原料8-羟基喹啉经亚硝化和氧化两步合成而得,制备方法参见《8-羟基喹啉定向硝化的研究》,淮阴师范学院学报,2005年2月第4卷第1期。
100.实施例1硝羟喹啉的a型结晶的制备例一
101.称取硝羟喹啉50mg,于50℃,逐渐加入甲醇(6ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至室温,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄色长针状固体约40mg,收率:80%。
102.所得固体的x射线粉末衍射图谱见图1,x射线粉末衍射数据见下表1,且被定义为a型结晶。
103.表1晶型a的xrpd峰值与强度列表
104.[0105][0106]
称取大约5mg该结晶样品于坩埚中,氮气保护,从30℃升温至250℃,升温速率为20℃/min,250℃保持1分钟。其tga图谱见图2,其显示该晶型从室温至120℃无失重,但之后有升华现象。
[0107]
称取大约5mg粉末样品放置在一个封闭的铝坩埚中,坩埚盖上扎一针孔。氮气保护,从30℃升温到300℃进行差示热量扫描,300℃保持1分钟。升温速率为20℃/min。其dsc图谱见图3,其显示在180.82℃处有一个明显的吸热峰。
[0108]
实施例2硝羟喹啉的a型结晶的制备例二
[0109]
称取硝羟喹啉50mg,于60℃,逐渐加入乙腈(2.5ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至-20℃,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄色长针状固体约35mg,收率:70%。
[0110]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0111]
实施例3硝羟喹啉的a型结晶的制备例三
[0112]
称取硝羟喹啉50mg,于55℃,逐渐加入异丙醇(15ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至-20℃,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄色固体约35mg,收率:70%。
[0113]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0114]
实施例4硝羟喹啉的a型结晶的制备例四
[0115]
称取硝羟喹啉50mg,于60℃,逐渐加入乙酸乙酯(2.5ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至-10℃,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄绿色长针状固体约40mg,收率:80%。
[0116]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0117]
实施例5硝羟喹啉的a型结晶的制备例五
[0118]
称取硝羟喹啉50mg,于60℃,逐渐加入丙酮(2ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至-20℃,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄色长针状固体约40mg,收率:80%。
[0119]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0120]
实施例6硝羟喹啉的a型结晶的制备例六
[0121]
称取硝羟喹啉50mg,于50℃,逐渐加入甲基叔丁基醚(10ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至室温,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄色絮状固体约40mg,收率:80%。
[0122]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0123]
实施例7硝羟喹啉的a型结晶的制备例七
[0124]
称取硝羟喹啉50mg,于55℃,逐渐加入甲苯(4ml),直至使得硝羟喹啉完全溶解至澄清溶液。然后,将溶液降至-20℃,搅拌0.5h,离心(eppendof centrifuge 5415,4000r/min,5min)分离并收集湿固体,于40℃减压真空干燥,得黄色长针状固体约40mg,收率:80%。
[0125]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0126]
实施例8硝羟喹啉的a型结晶的制备例八
[0127]
称取硝羟喹啉50mg,于室温,加入二甲亚砜(0.6ml)使其完全溶解至澄清,然后,逐渐加入水(2.4ml),使固体析出,离心分离并收集湿固体,于40℃减压真空干燥,得黄色细小针状固体约45mg,收率:90%。
[0128]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0129]
实施例9硝羟喹啉的a型结晶的制备例九
[0130]
称取硝羟喹啉50mg,于室温,加入n,n-二甲基甲酰胺(0.6ml)使其完全溶解至澄清,然后,逐渐加入水(3.6ml),使固体析出,离心分离并收集湿固体,于40℃减压真空干燥,得黄色细小针状固体约45mg,收率:90%。
[0131]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0132]
实施例10硝羟喹啉的a型结晶的制备例十
[0133]
称取硝羟喹啉50mg,于室温,加入n-甲基吡咯烷酮(0.5ml)使其完全溶解至澄清,然后,逐渐加入水(1.5ml),使固体析出,离心分离并收集湿固体,于40℃减压真空干燥,得黄色细小针状固体约45mg,收率:90%。
[0134]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0135]
实施例11硝羟喹啉的a型结晶的制备例十一
[0136]
称取硝羟喹啉50mg,于室温,加入乙酸(5ml)使其完全溶解至澄清,然后,逐渐加入水(10ml),使固体析出,离心分离并收集湿固体,于40℃减压真空干燥,得黄色细小针状固体约45mg,收率:90%。
[0137]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0138]
实施例12硝羟喹啉的a型结晶的制备例十二
[0139]
称取硝羟喹啉50mg,于室温,加入丙酮(4ml)使其完全溶解至澄清,然后,逐渐加入水(20ml),使固体析出,离心分离并收集湿固体,于40℃减压真空干燥,得黄色细小针状固体约45mg,收率:90%。
[0140]
其xrpd图谱经研究比对,确定产物为a型结晶。
[0141]
实施例13硝羟喹啉的a型结晶的制备例十三
[0142]
称取硝羟喹啉50mg,于室温,加入甲醇(10ml)使其完全溶解至澄清,然后,逐渐加入水(50ml),使固体析出,离心分离并收集湿固体,于40℃减压真空干燥,得黄色细小针状固体约45mg。
[0143]
其xrpd图谱经研究比对,确定产物为a型结晶,收率:90%。
[0144]
测试例1本发明硝羟喹啉a型结晶的吸湿性试验
[0145]
称取实施例8制得的硝羟喹啉a型结晶1-5mg,置动态水分吸附仪中,直接测定。设置仪器参数,样品测试温度:t=25℃,平衡时间:dm/dt:0.01%/min,相对湿度变化范围:0%-95%-0%;rh(%)测试每步湿度变化:5%。
[0146]
吸湿性的评价标准如下表2所示。
[0147]
表2吸湿性分类
[0148]
吸湿性分类水份吸附标准
*
易潮解的吸附足够多的水份成液体状非常吸湿的w%≥15%吸湿的w%≥2%轻微吸湿的w%≥0.2%不吸湿的w%<0.2%
[0149]
*在25
±
1℃和80
±
2%rh(欧洲药典6.0)
[0150]
实施例8的a型结晶的dvs试验结果如图4所示,其中显示水分吸附值w%<0.2%,说明本发明a型结晶是不吸湿的,具有良好的稳定性。
[0151]
测试例2本发明硝羟喹啉a型结晶的稳定性试验
[0152]
称取实施例8制得的硝羟喹啉a型结晶分别置光照(4500
±
500lux)、高温(60℃)、高湿(rh90%
±
5%)条件下影响因素考察5天、10天,进行x-射线粉末衍射(xrpd)检测,考察其晶型是否发生改变。
[0153]
晶型稳定性影响因素试验结果如下表3所示。
[0154]
表3晶型稳定性影响因素试验结果
[0155][0156]
本发明a型晶型,经光照、高温、高湿因素10天考察后,晶型未发生改变,具有良好的稳定性。
[0157]
虽然以上描述了本发明的具体实施方式,但是本领域的技术人员应当理解,这仅是举例说明,本发明的保护范围是由所附权利要求书限定的。本领域的技术人员在不背离本发明的原理和实质的前提下,可以对这些实施方式做出多种变更或修改,但这些变更和修改均落入本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1