一种不对称扩增多个靶核酸的方法
1.本申请涉及核酸分子的多重、不对称扩增。特别地,本申请提供了一种同时、不对称扩增样品中的一个或多个靶核酸的方法,所述方法能够同时不对称扩增样品中存在的多种靶核酸,能够同时产生大量的单链产物。
背景技术:
2.不对称pcr,由gyllensten等人(proc.natl.acad.sci.usa 1988,85:7652-7656)首次描述,是指利用不等量的一对引物来产生大量单链dna(ssdna)的方法。不对称pcr产生的单链dna可以用于测序,用作探针,或在实时pcr、基因芯片检测、探针熔解曲线分析中改善检测信号。然而,传统的不对称pcr通常需要精心的优化,以最大限度地产生特定的单链产物和最小化非特异性扩增。当需要同时不对称扩增多个靶核酸序列时,由于引物对的增加,导致引物二聚体等非特异性扩增的增加,更增加了设计和优化的难度。
3.在传统的不对称pcr扩增中,由于限制性引物浓度降低,导致其熔点低于pcr反应的退火温度,这进而导致了不对称pcr扩增的低效性。针对此,sanchez等人(proc.natl.acad.sci.usa 2004,101:1933-1938)提出late-pcr(linear-after-the-exponential-pcr),其中,在引物设计时增加了引物的实际使用浓度作为影响引物熔点(t
m
)值的因素,以期提高低浓度限制性引物的t
m
值,使其高于退火温度,从而提高不对称pcr的扩增效率,以产生大量的单链产物。该方法解决了不对称pcr扩增效率低的问题,使不对称pcr更易于优化。然而,该方法未能解决引物对增加导致的非特异性扩增增加的问题,仍然很难实现多重不对称pcr扩增。
4.brownie等人(nucleic acids research 1997,26:3235-3241)描述了一种同源标签辅助的无引物二聚体系统(homo-tag assisted non-dimer system,hand系统)。在该系统中,在靶特异的上游引物和下游引物的5'端均加上同样的标签序列,形成加尾/加标签的靶特异性引物。在pcr扩增时,首先由低浓度的加尾/加标签的特异性引物在较低的退火温度下启动初始pcr扩增;几个循环之后,在升高的退火温度下,由一条高浓度的通用引物对初始pcr扩增的扩增产物进行后续扩增。由于特异性引物均含有同样的标签序列,由初始pcr扩增产生的所有产物(包括引物二聚体)的末端都具有互补的标签序列。引物二聚体等小片段产物的单链由于局部浓度较高容易自身退火形成稳定的“锅柄(pan-handle)”结构,阻止通用引物进一步的退火,从而抑制引物二聚体的扩增。通过结合采用低浓度的加同源标签的靶特异性引物和高浓度的通用引物,hand系统能够有效抑制引物二聚体的扩增,实现多重pcr的有效扩增,并保持较高的扩增效率和检测灵敏度。然而,使用hand系统的pcr扩增是对称扩增,无法产生单链产物,这导致hand系统在基因芯片、探针熔解曲线分析等技术领域的应用受到了限制。
5.因此,需要开发新的能够同时不对称扩增多个靶核酸的方法,其能够同时实现有效的多重pcr扩增和不对称pcr扩增,满足临床上对多个靶核酸同时进行扩增和检测的要求。
技术实现要素:
6.在本申请中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员所通常理解的含义。并且,本文中所用的核酸化学实验室操作步骤均为相应领域内广泛使用的常规步骤。同时,为了更好地理解本发明,下面提供相关术语的定义和解释。
7.如本文中所使用的,术语“靶核酸序列”、“靶核酸”和“靶序列”是指待检测的目标核酸或其序列。在本申请中,术语“靶核酸序列”、“靶核酸”和“靶序列”具有相同的含义,并且可互换使用。
8.如本文中所使用的,术语“特异于靶核酸的序列”和“靶特异性序列”是指,在允许核酸杂交、退火或扩增的条件下,能够与靶核酸选择性/特异性杂交或退火的序列,其包含与靶核酸序列互补的序列。在本申请中,术语“特异于靶核酸的序列”和“靶特异性序列”具有相同的含义,并且可互换使用。易于理解的是,特异于靶核酸的序列或靶特异性序列对于靶核酸是特异性的。换言之,在允许核酸杂交、退火或扩增的条件下,特异于靶核酸的序列或靶特异性序列仅与特定的靶核酸杂交或退火,而不与其他的核酸序列杂交或退火。例如,在本申请中,“特异于靶核酸的正向核苷酸序列”意指,在允许核酸杂交、退火或扩增的条件下,能够与靶核酸选择性/特异性杂交或退火的正向核苷酸序列,其包含与靶核酸互补的序列。
9.如本文中所使用的,术语“互补”意指,两条核酸序列能够根据碱基配对原则(waston-crick原则)在彼此之间形成氢键,并由此形成双链体。在本申请中,术语“互补”包括“实质上互补”和“完全互补”。如本文中所使用的,术语“完全互补”意指,一条核酸序列中的每一个碱基都能够与另一条核酸链中的碱基配对,而不存在错配或缺口。如本文中所使用的,术语“实质上互补”意指,一条核酸序列中的大部分碱基都能够与另一条核酸链中的碱基配对,其允许存在错配或缺口(例如,一个或数个核苷酸的错配或缺口)。通常,在允许核酸杂交、退火或扩增的条件下,“互补”(例如实质上互补或完全互补)的两条核酸序列将选择性地/特异性地发生杂交或退火,并形成双链体。相应地,术语“不互补”意指,两条核酸序列在允许核酸杂交、退火或扩增的条件下不能发生杂交或退火,无法形成双链体。如本文中所使用的,术语“不能完全互补”意指,一条核酸序列中的碱基不能够与另一条核酸链中的碱基完全配对,至少存在一个错配或缺口。
10.如本文中所使用的,术语“杂交”和“退火”意指,互补的单链核酸分子形成双链核酸的过程。在本申请中,“杂交”和“退火”具有相同的含义,并且可互换使用。通常,完全互补或实质上互补的两条核酸序列可发生杂交或退火。两条核酸序列发生杂交或退火所需要的互补性取决于所使用的杂交条件,特别是温度。
11.如本文中所使用的,“允许核酸杂交的条件”具有本领域技术人员通常理解的含义,并且可通过常规的方法来确定。例如,具有互补序列的两条核酸分子可在合适的杂交条件下发生杂交。此类杂交条件可涉及下列因素:温度,杂交缓冲液的ph值、成分和离子强度等,并且可根据互补的两条核酸分子的长度和gc含量来确定。例如,当互补的两条核酸分子的长度相对较短和/或gc含量相对较低时,可采用低严紧的杂交条件。当互补的两条核酸分子的长度相对较长和/或gc含量相对较高时,可采用高严紧的杂交条件。此类杂交条件是本领域技术人员熟知的,并且可参见例如joseph sambrook,et al.,molecular cloning,a laboratory manual,cold spring harbor laboratory press,cold spring harbor,n.y.
(2001);和m.l.m.anderson,nucleic acid hybridization,springer-verlag new york inc.n.y.(1999)。在本申请中,“杂交”和“退火”具有相同的含义,并且可互换使用。相应地,表述“允许核酸杂交的条件”和“允许核酸退火的条件”也具有相同的含义,并且可互换使用。
12.如本文中所使用的,表述“允许核酸扩增的条件”具有本领域技术人员通常理解的含义,其是指,允许核酸聚合酶(例如dna聚合酶)以一条核酸链为模板合成另一条核酸链,并形成双链体的条件。此类条件是本领域技术人员熟知的,并且可涉及下列因素:温度,杂交缓冲液的ph值、成分、浓度和离子强度等。可通过常规方法来确定合适的核酸扩增条件(参见例如joseph sambrook,et al.,molecular cloning,a laboratory manual,cold spring harbor laboratory press,cold spring harbor,n.y.(2001))。在本发明的方法中,“允许核酸扩增的条件”优选地为核酸聚合酶(例如dna聚合酶)的工作条件。
13.如本文中所使用的,表述“允许核酸聚合酶进行延伸反应的条件”具有本领域技术人员通常理解的含义,其是指,允许核酸聚合酶(例如dna聚合酶)以一条核酸链为模板延伸另一条核酸链(例如引物或探针),并形成双链体的条件。此类条件是本领域技术人员熟知的,并且可涉及下列因素:温度,杂交缓冲液的ph值、成分、浓度和离子强度等等。可通过常规方法来确定合适的核酸扩增条件(参见例如joseph sambrook,et al.,molecular cloning,a laboratory manual,cold spring harbor laboratory press,cold spring harbor,n.y.(2001))。在本发明的方法中,“允许核酸聚合酶进行延伸反应的条件”优选地为核酸聚合酶(例如dna聚合酶)的工作条件。在本申请中,表述“允许核酸聚合酶进行延伸反应的条件”和“允许核酸延伸的条件”具有相同的含义,并且可互换使用。
14.各种酶的工作条件可由本领域技术人员通过常规方法确定,并且通常可涉及下列因素:温度,缓冲液的ph值,成分,浓度,离子强度等。备选地,可使用酶的制造商所推荐的条件。
15.如本文中所使用的,术语“核酸变性”具有本领域技术人员通常理解的含义,其是指,双链核酸分子解离为单链的过程。表述“允许核酸变性的条件”是指,使得双链核酸分子解离为单链的条件。此类条件可由本领域技术人员常规地确定(参见例如joseph sambrook,et al.,molecular cloning,a laboratory manual,cold spring harbor laboratory press,cold spring harbor,n.y.(2001))。例如,可通过加热,碱处理,尿素处理,酶促方法(例如使用解旋酶的方法)等常规技术来使核酸变性。在本申请中,优选地,在加热的条件下使核酸变性。例如,可通过加热至80-105℃,从而使核酸变性。
16.如本文中所使用的,术语“pcr反应”具有本领域技术人员通常理解的含义,其是指使用核酸聚合酶和引物来扩增靶核酸的反应(聚合酶链式反应)。如本文中所使用的,术语“多重扩增”是指,在同一个反应体系中对多个靶核酸进行扩增。如本文中所使用的,术语“不对称扩增”是指,对靶核酸进行扩增所获得的扩增产物中,两条互补的核酸链的量不相同,一条核酸链的量大于另一条核酸链。
17.如本文中所使用的,并且如本领域技术人员通常理解的,术语“正向”和“反向”仅仅是为了便于描述和区分一个引物对中的两条引物;它们是相对而言的,并不具有特别的含义。
18.如本文中所使用的,术语“荧光探针”是指携带荧光基团、且能够产生荧光信号的
一段寡核苷酸。
19.如本文中所使用的,术语“熔解曲线分析”具有本领域技术人员通常理解的含义,其是指,通过测定双链核酸分子的熔解曲线来分析双链核酸分子存在或其身份(identity)的方法,其通常用于评估双链核酸分子在加热过程中的解离特征。用于进行熔解曲线分析的方法是本领域技术人员熟知的(参见例如the journal of molecular diagnostics 2009,11(2):93-101)。在本申请中,术语“熔解曲线分析”和“熔解分析”具有相同的含义,并且可互换使用。
20.在本申请的某些优选实施方案中,可通过使用标记有报告基团和淬灭基团的检测探针来进行熔解曲线分析。简言之,在环境温度下,检测探针能够通过碱基配对作用与其互补序列形成双链体。在此情况下,检测探针上的报告基团(例如荧光基团)和淬灭基团彼此分离,淬灭基团无法吸收报告基团发出的信号(例如荧光信号),此时,能够检测到最强的信号(例如荧光信号)。随着温度的升高,双链体的两条链开始解离(即,检测探针逐渐从其互补序列上解离),并且解离下的检测探针呈单链自由卷曲状态。在此情况下,解离下的检测探针上的报告基团(例如荧光基团)和淬灭基团互相靠近,由此报告基团(例如荧光基团)发出的信号(例如荧光信号)被淬灭基团所吸收。因此,随着温度的升高,所检测到信号(例如荧光信号)逐渐变弱。当双链体的两条链完全解离时,所有的检测探针均呈单链自由卷曲状态。在此情况下,所有的检测探针上的报告基团(例如荧光基团)发出的信号(例如荧光信号)都被淬灭基团所吸收。因此,基本上无法检测到报告基团(例如荧光基团)发出的信号(例如荧光信号)。因此,对包含检测探针的双链体在升温或降温过程中发出的信号(例如荧光信号)进行检测,就能观察到检测探针与其互补序列的杂交和解离过程,形成信号强度随着温度变化而变化的曲线。进一步,对所获得的曲线进行求导分析,可获得以信号强度变化速率为纵坐标,温度为横坐标的曲线(即,该双链体的熔解曲线)。该熔解曲线中的峰即为熔解峰,其所对应的温度即为所述双链体的熔点(t
m
值)。通常而言,检测探针与互补序列的匹配程度越高(例如,错配的碱基越少,配对的碱基越多),那么双链体的t
m
值就越高。因此,通过检测双链体的t
m
值,可确定双链体中与检测探针互补的序列的存在和身份。在本文中,术语“熔解峰”、“熔点”和“t
m
值”具有相同的含义,并且可互换使用。
21.扩增方法
22.在一个方面,本发明提供了一种扩增样品中的一种或多种靶核酸的方法,其包括,
23.(1)提供含有一种或多种靶核酸的样品;和,提供第一通用引物和第二通用引物,以及,针对每一种待扩增的靶核酸,提供一个靶特异性引物对;其中,
24.所述第一通用引物包含第一通用序列;
25.所述第二通用引物包含第二通用序列,所述第二通用序列包含第一通用序列且在第一通用序列的3'端额外包含至少一个核苷酸;
26.所述靶特异性引物对能够扩增所述靶核酸,并且包含一个正向引物和一个反向引物,其中,所述正向引物包含第一通用序列和特异于所述靶核酸的正向核苷酸序列,且所述正向核苷酸序列位于第一通用序列的3'端;所述反向引物包含第二通用序列和特异于所述靶核酸的反向核苷酸序列,且所述反向核苷酸序列位于第二通用序列的3'端;并且,第二通用序列不能与所述正向引物的互补序列完全互补;
27.(2)在允许核酸扩增的条件下,使用所述第一通用引物、所述第二通用引物以及所
述靶特异性引物对,通过pcr反应扩增样品中的靶核酸。
28.在本发明的方法中,正向引物和反向引物分别包含特异于所述靶核酸的正向核苷酸序列和反向核苷酸序列,由此,在pcr反应过程中,靶特异性引物对(正向引物和反向引物)将退火至靶核酸,并启动pcr扩增,产生初始扩增产物,其包含分别与正向引物和反向引物互补的两条核酸链(核酸链a和核酸链b)。进一步,由于正向引物和第一通用引物均包含第一通用序列,因此,与正向引物互补的核酸链a也能够与第一通用引物互补。类似地,与反向引物互补的核酸链b也能够与第二通用引物互补。
29.因此,随着pcr反应的进行,第一通用引物和第二通用引物将分别退火至初始扩增产物的核酸链a和核酸链b,并进一步启动pcr扩增。在该过程中,由于反向引物/第二通用引物包含第一通用序列,因此,第一通用引物不仅能够退火至核酸链a(与正向引物/第一通用引物互补的核酸链)并合成其互补链,而且能够退火至核酸链b(与反向引物/第二通用引物互补的核酸链)并合成其互补链。也即,第一通用引物可以同时扩增初始扩增产物的核酸链a和核酸链b。与此同时,第二通用引物在第一通用序列的3'端包含额外的核苷酸,因此,虽然第二通用引物也可能退火至核酸链a(与正向引物/第一通用引物互补的核酸链,其具有与正向引物互补的序列),但是其与核酸链a在3'端是不匹配的(即,在3'端不能完全互补)。由此,在扩增过程中,第二通用引物将优先退火至核酸链b(与反向引物/第二通用引物互补的核酸链)并合成其互补链,而基本上不能延伸合成核酸链a(与第一正向引物/第一通用引物互补的核酸链)的互补链。
30.因此,随着pcr扩增的进行,核酸链a的互补链(核酸链b)的合成效率将显著低于核酸链b的互补链(核酸链a),导致核酸链b的互补链(核酸链a)被大量合成和扩增,而核酸链a的互补链(核酸链b)的合成和扩增受到抑制,从而产生大量单链产物(核酸链a,其含有与正向引物/第一通用引物互补的序列以及反向引物/第二通用引物的序列),实现了对靶核酸的不对称扩增。因此,在某些优选的实施方案中,本发明的方法能够不对称扩增样品中的一种或多种靶核酸。
31.另外,由于正向引物和反向引物均含有第一通用序列,因此,在pcr反应过程中,因正向引物和反向引物的非特异性扩增而形成的引物二聚体在变性后将产生其5'端和3'端包含彼此互补的反向序列的单链核酸,该单链核酸在退火阶段容易自身退火,形成稳定的锅柄结构,阻止第一通用引物和第二通用引物对该单链核酸的退火和延伸,从而抑制引物二聚体的进一步扩增。因此,在本发明的方法中,引物二聚体的非特异性扩增能够被有效抑制。由此,本发明的方法特别适合用于进行多重扩增。例如,在本发明的方法中,可以将第一通用引物和第二通用引物与一个或多个靶特异性引物对组合使用,实现对一个或多个靶核酸的多重扩增。
32.因此,在某些优选的实施方案中,本发明的方法能够同时扩增1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶核酸,例如至少2个,至少3个,至少4个,至少5个,至少8个,至少10个,至少12个,至少15个,至少18个,至少20个,至少25个,至少30个,至少40个,至少50个,或更多个靶核酸。在某些优选的实施方案中,本发明的方法能够同时并且不对称扩增1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶核酸。在此类实施方案中,相应地,在步骤(1)中针对每一种待扩增的靶核酸,提供一个靶特异性引物对。因此,在此类实施方案中,在步骤(1)中提供1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶特异性引物
对,例如至少2个,至少3个,至少4个,至少5个,至少8个,至少10个,至少12个,至少15个,至少18个,至少20个,至少25个,至少30个,至少40个,至少50个,或更多个靶特异性引物对。
33.易于理解,对于不同的靶核酸,可以使用不同的正向引物和反向引物。然而,当不同的靶核酸之间存在序列相似性时,不同的靶特异性引物对可能具有相同的正向引物或反向引物。
34.为了便于进行多重不对称扩增且有效抑制引物二聚体的非特异性扩增,在某些优选的实施方案中,所述第一通用引物和第二通用引物的工作浓度高于所述正向引物和反向引物的工作浓度。在某些优选的实施方案中,所述第一通用引物和第二通用引物的工作浓度比所述正向引物和反向引物的工作浓度高至少1倍,至少2倍,至少3倍,至少4倍,至少5倍,至少8倍,至少10倍,至少12倍,至少15倍,至少18倍,至少20倍,至少25倍,至少30倍,至少40倍,至少50倍或更多倍。在某些优选的实施方案中,所述第一通用引物和第二通用引物的工作浓度比所述正向引物和反向引物的工作浓度高1-5倍,5-10倍,10-15倍,15-20倍,20-50倍或更多倍。
35.在本发明的方法中,所述第一通用引物和第二通用引物的工作浓度可以相同或者不同。在某些优选的实施方案中,所述第一通用引物和第二通用引物的工作浓度是相同的。在某些优选的实施方案中,所述第一通用引物和第二通用引物的工作浓度是不同的。如上文所详细论述的,在本发明的方法中,通过核酸链a和b与第一和第二通用引物的匹配性差异来实现不对称扩增。因此,第一通用引物相比于第二通用引物的相对浓度是可以变化的。在某些优选的实施方案中,所述第一通用引物和第二通用引物的工作浓度是相同的。在某些优选的实施方案中,所述第一通用引物的工作浓度高于第二通用引物。在某些优选的实施方案中,所述第一通用引物的工作浓度低于第二通用引物。如上文所详细论述的,本发明的方法可用于实现靶核酸的不对称扩增。在某些情况下,更高的扩增不对称性可能是有利的。因此,在某些优选的实施方案中,还可以调整第一通用引物和第二通用引物的比例,使得第一通用引物的工作浓度低于第二通用引物,以进一步增强扩增的不对称性,更好地富集单链产物。
36.在本发明的方法中,所述正向引物和反向引物的工作浓度可以相同或者不同。在某些优选的实施方案中,所述正向引物和反向引物的工作浓度是相同的。在某些优选的实施方案中,所述正向引物和反向引物的工作浓度是不同的。在某些优选的实施方案中,所述正向引物的工作浓度低于所述反向引物的工作浓度。在某些优选的实施方案中,所述正向引物的工作浓度高于所述反向引物的工作浓度。
37.在某些优选的实施方案中,所述第一通用引物由第一通用序列组成。在某些优选的实施方案中,所述第一通用引物还包含额外的序列,其位于第一通用序列的5'端。在某些优选的实施方案中,所述额外的序列包含一个或多个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。在本申请中,所述第一通用引物用于进行pcr扩增,因此优选地,第一通用序列位于或构成所述第一通用引物的3'部分。
38.在本申请的实施方案中,所述第一通用引物可以是任意的长度,只要其能够进行pcr反应即可。例如,所述第一通用引物长度可以为5-50nt,例如5-15nt,15-20nt,20-30nt,30-40nt,或40-50nt。
39.在本申请的某些实施方案中,所述第一通用引物(或其任何组成成分)可以包含或者由天然存在的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸),经修饰的核苷酸,非天然的核苷酸,或其任何组合组成。在某些优选的实施方案中,第一通用引物(或其任何组成成分)包含或者由天然的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸)组成。在某些优选的实施方案中,第一通用引物(或其任何组成成分)包含经修饰的核苷酸,例如经修饰的脱氧核糖核苷酸或核糖核苷酸,例如5-甲基胞嘧啶或5-羟甲基胞嘧啶。在某些优选的实施方案中,第一通用引物(或其任何组成成分)包含非天然的核苷酸,例如脱氧次黄嘌呤,肌苷,1-(2'-脱氧-β-d-呋喃核糖基)-3-硝基吡咯,5-硝基吲哚或锁核酸(lna)。
40.在某些优选的实施方案中,所述第二通用引物由第二通用序列组成。在某些优选的实施方案中,所述第二通用引物还包含额外的序列,其位于第二通用序列的5'端。在某些优选的实施方案中,所述额外的序列包含一个或多个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。在本申请中,所述第二通用引物用于进行pcr扩增,因此优选地,第二通用序列位于或构成所述第二通用引物的3'部分。
41.在某些优选的实施方案中,所述第二通用序列包含第一通用序列且在第一通用序列的3'端额外包含至少一个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。
42.在本申请的实施方案中,所述第二通用引物可以是任意的长度,只要其能够进行pcr反应并满足如上所定义的条件即可。例如,所述第二通用引物长度可以为8-50nt,例如8-15nt,15-20nt,20-30nt,30-40nt,或40-50nt。在本申请的实施方案中,第二通用序列的长度大于第一通用序列的长度。
43.在本申请的某些实施方案中,所述第二通用引物(或其任何组成成分)可以包含或者由天然存在的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸),经修饰的核苷酸,非天然的核苷酸,或其任何组合组成。在某些优选的实施方案中,第二通用引物(或其任何组成成分)包含或者由天然的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸)组成。在某些优选的实施方案中,第二通用引物(或其任何组成成分)包含经修饰的核苷酸,例如经修饰的脱氧核糖核苷酸或核糖核苷酸,例如5-甲基胞嘧啶或5-羟甲基胞嘧啶。在某些优选的实施方案中,第二通用引物(或其任何组成成分)包含非天然的核苷酸,例如脱氧次黄嘌呤,肌苷,1-(2'-脱氧-β-d-呋喃核糖基)-3-硝基吡咯,5-硝基吲哚或锁核酸(lna)。
44.在本申请的某些实施方案中,在步骤(1)中提供至少1个靶特异性引物对,例如,至少2个,至少3个,至少4个,至少5个,至少8个,至少10个,至少12个,至少15个,至少18个,至少20个,至少25个,至少30个,至少40个,至少50个,或更多个靶特异性引物对。在某些优选的实施方案中,在步骤(1)中提供1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶特异性引物对。在某些优选的实施方案中,所述方法能够同时扩增1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶核酸。在某些优选的实施方案中,所述方法能够同时并且不对称扩增1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶核酸。
45.例如,在某些优选的实施方案中,本发明的方法可用于扩增样品中的2种靶核酸,其中,在步骤(1)中提供第一通用引物,第二通用引物,第一靶特异性引物对和第二靶特异性引物对;其中,第一通用引物和第二通用引物如上文所定义,并且,第一靶特异性引物对
包含能够扩增第一靶核酸的第一正向引物和第一反向引物,第二靶特异性引物对包含能够扩增第二靶核酸的第二正向引物和第二反向引物;其中,第一正向引物、第一反向引物、第二正向引物和第二反向引物如上文所定义。类似地,在某些优选的实施方案中,本发明的方法可用于扩增样品中的3种或更多种靶核酸,其中,在步骤(1)中提供第一通用引物,第二通用引物,以及能够扩增所述3种或更多种靶核酸的3种或更多种靶特异性引物对。
46.在某些优选的实施方案中,在所述正向引物中,所述正向核苷酸序列直接连接至第一通用序列的3'端。在某些优选的实施方案中,在所述正向引物中,所述正向核苷酸序列通过核苷酸连接体连接至第一通用序列的3'端。在某些优选的实施方案中,所述正向引物从5'至3'包含或由第一通用序列和正向核苷酸序列组成。在某些优选的实施方案中,所述正向引物从5'至3'包含或由第一通用序列、核苷酸连接体和正向核苷酸序列组成。在某些优选的实施方案中,所述核苷酸连接体包含一个或多个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。
47.在某些优选的实施方案中,所述正向引物还包含额外的序列,其位于第一通用序列的5'端。因此,在某些优选的实施方案中,所述正向引物从5'至3'包含或由额外的序列、第一通用序列和正向核苷酸序列组成。在某些优选的实施方案中,所述正向引物从5'至3'包含或由额外的序列、第一通用序列、核苷酸连接体和正向核苷酸序列组成。在某些优选的实施方案中,所述额外的序列包含一个或多个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。
48.在本申请中,所述正向引物用于对靶核酸进行pcr扩增,因此优选地,正向核苷酸序列位于或构成所述正向引物的3'部分。
49.在本申请的实施方案中,正向核苷酸序列不受其长度的限制,只要其能够与靶核酸序列特异性杂交并扩增靶核酸即可。例如,正向核苷酸序列的长度可以为10-100nt,例如10-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt,80-90nt,90-100nt。
50.在本申请的实施方案中,正向引物不受其长度的限制,只要其满足如上所定义的条件即可。例如,正向引物的长度可以为15-150nt,例如15-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt,80-90nt,90-100nt,100-110nt,110-120nt,120-130nt,130-140nt,140-150nt。
51.在本申请的某些实施方案中,正向引物(或其任何组成成分)可以包含或者由天然存在的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸),经修饰的核苷酸,非天然的核苷酸,或其任何组合组成。在某些优选的实施方案中,正向引物(或其任何组成成分)包含或者由天然的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸)组成。在某些优选的实施方案中,正向引物(或其任何组成成分)包含经修饰的核苷酸,例如经修饰的脱氧核糖核苷酸或核糖核苷酸,例如5-甲基胞嘧啶或5-羟甲基胞嘧啶。在某些优选的实施方案中,正向引物(或其任何组成成分)包含非天然的核苷酸,例如脱氧次黄嘌呤,肌苷,1-(2'-脱氧-β-d-呋喃核糖基)-3-硝基吡咯,5-硝基吲哚或锁核酸(lna)。
52.在某些优选的实施方案中,在所述反向引物中,所述反向核苷酸序列直接连接至第二通用序列的3'端。在某些优选的实施方案中,在所述反向引物中,所述反向核苷酸序列通过核苷酸连接体连接至第二通用序列的3'端。在某些优选的实施方案中,所述反向引物从5'至3'包含或由第二通用序列和反向核苷酸序列组成。在某些优选的实施方案中,所述
反向引物从5'至3'包含或由第二通用序列、核苷酸连接体和反向核苷酸序列组成。在某些优选的实施方案中,所述核苷酸连接体包含一个或多个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。
53.在某些优选的实施方案中,所述反向引物还包含额外的序列,其位于第二通用序列的5'端。因此,在某些优选的实施方案中,所述反向引物从5'至3'包含或由额外的序列、第二通用序列和反向核苷酸序列组成。在某些优选的实施方案中,所述反向引物从5'至3'包含或由额外的序列、第二通用序列、核苷酸连接体和反向核苷酸序列组成。在某些优选的实施方案中,所述额外的序列包含一个或多个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸。
54.在本申请中,所述反向引物用于对靶核酸进行pcr扩增,因此优选地,反向核苷酸序列位于或构成所述反向引物的3'部分。
55.在本申请的实施方案中,反向核苷酸序列不受其长度的限制,只要其能够与靶核酸序列特异性杂交并扩增靶核酸即可。例如,反向核苷酸序列的长度可以为10-100nt,例如10-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt,80-90nt,90-100nt。
56.在本申请的实施方案中,反向引物不受其长度的限制,只要其满足如上所定义的条件即可。例如,反向引物的长度可以为15-150nt,例如15-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt,80-90nt,90-100nt,100-110nt,110-120nt,120-130nt,130-140nt,140-150nt。
57.在本申请的某些实施方案中,反向引物(或其任何组成成分)可以包含或者由天然存在的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸),经修饰的核苷酸,非天然的核苷酸,或其任何组合组成。在某些优选的实施方案中,反向引物(或其任何组成成分)包含或者由天然的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸)组成。在某些优选的实施方案中,反向引物(或其任何组成成分)包含经修饰的核苷酸,例如经修饰的脱氧核糖核苷酸或核糖核苷酸,例如5-甲基胞嘧啶或5-羟甲基胞嘧啶。在某些优选的实施方案中,反向引物(或其任何组成成分)包含非天然的核苷酸,例如脱氧次黄嘌呤,肌苷,1-(2'-脱氧-β-d-呋喃核糖基)-3-硝基吡咯,5-硝基吲哚或锁核酸(lna)。
58.在本申请的实施方案中,第二通用序列不能与所述正向引物的互补序列完全互补。在某些优选的实施方案中,第二通用序列中位于3'末端的至少一个核苷酸,例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸,例如1、2、3、4、5、6、7、8、9或10个核苷酸不能与所述正向引物的互补序列互补。由此,在pcr反应过程中,第二通用序列/第二通用引物即使能够退火至与正向引物互补的核酸链(核酸链a)上,也基本上不能延伸合成所述核酸链的互补链。
59.在本发明的方法中,样品可以是任何包含核酸的样品。例如,在某些优选的实施方案中,样品包含或是dna(例如基因组dna或cdna)。在某些优选的实施方案中,样品包含或者是rna(例如mrna)。在某些优选的实施方案中,样品包含或者是核酸的混合物(例如dna的混合物,rna的混合物,或者dna和rna的混合物)。
60.在本发明的方法中,待扩增的靶核酸不受限于其序列组成或长度。例如,所述靶核酸可以是dna(例如基因组dna或cdna)或rna分子(例如mrna)。此外,待扩增的靶核酸可以是单链的或双链的。
61.当所述样品或靶核酸为mrna时,优选地,在进行本发明的方法之前,进行逆转录反应,以获得与所述mrna互补的cdna。关于逆转录反应的详细描述可参见例如,joseph sam-brook,et al.,molecular cloning,alaboratory manual,cold spring harbor laboratory press,cold springharbor,n.y.(2001)。
62.在本发明的方法中,所述样品或靶核酸可获自任何来源,包括但不限于原核生物,真核生物(例如原生动物,寄生虫,真菌,酵母,植物,动物包括哺乳动物和人类)或病毒(例如herpes病毒,hiv,流感病毒,eb病毒,肝炎病毒,脊髓灰质炎病毒等)或类病毒。所述样品或靶核酸还可以是任何形式的核酸,例如基因组核酸,人工分离或片段化的核酸,合成的核酸等。
63.在本发明的方法中,可以使用任何核酸聚合酶(特别是模板依赖性核酸聚合酶)来进行pcr反应。在某些优选的实施方案中,所述核酸聚合酶为dna聚合酶。在某些优选的实施方案中,所述核酸聚合酶为热稳定的dna聚合酶。热稳定的dna聚合酶可获自各种细菌物种,例如但不限于,thermus aquaticus(taq),thermus thermophiles(tth),thermus filiformis,thermis flavus,thermococcus literalis,thermus antranildanii,thermus caldophllus,thermus chliarophilus,thermus flavus,thermus igniterrae,thermus lacteus,thermus oshimai,thermus ruber,thermus rubens,thermus scotoductus,thermus silvanus,thermus thermophllus,thermotoga maritima,thermotoga neapolitana,thermosipho africanus,thermococcus litoralis,thermococcus barossi,thermococcus gorgonarius,thermotoga maritima,thermotoga neapolitana,thermosiphoafricanus,pyrococcus woesei,pyrococcus horikoshii,pyrococcus abyssi,pyrodictium occultum,aquifexpyrophilus和aquifex aeolieus。特别优选地,所述dna聚合酶为taq聚合酶。
64.在某些优选的实施方案中,以三步法对靶核酸进行扩增。在此类实施方案中,每一轮的核酸扩增需要经过三个步骤:在第一温度下进行核酸变性,在第二温度下进行核酸退火,以及在第三温度下进行核酸延伸。在某些优选的实施方案中,以两步法对靶核酸进行扩增。在此类实施方案中,每一轮的核酸扩增需要经过两个步骤:在第一温度下进行核酸变性,以及在第二温度下进行核酸退火和延伸。适合于进行核酸变性、核酸退火和核酸延伸的温度可由本领域技术人员通过常规方法容易地确定(参见例如,joseph sambrook,et al.,molecular cloning,a laboratory manual,cold spring harbor laboratory press,cold spring harbor,n.y.(2001))。
65.在某些优选的实施方案中,本发明方法的步骤(1)-(2)可通过包含下述步骤(a)-(f)的方案来进行:
66.(a)提供含有一种或多种靶核酸的样品;和,提供第一通用引物和第二通用引物,以及,针对每一种待扩增的靶核酸,提供一个靶特异性引物对;其中,所述第一通用引物、第二通用引物和靶特异性引物对如上文所定义;
67.(b)将所述样品与所述第一通用引物、第二通用引物和靶特异性引物对,以及核酸聚合酶(例如,模板依赖性核酸聚合酶;例如dna聚合酶,特别是热稳定的dna聚合酶)混合;
68.(c)在允许核酸变性的条件下,温育前一步骤的产物;
69.(d)在允许核酸退火或杂交的条件下,温育前一步骤的产物;
70.(e)在允许核酸延伸的条件下,温育前一步骤的产物;和
71.(f)任选地,重复步骤(c)-(e)一次或多次。
72.在此类实施方案中,在步骤(c)中,样品中的所有核酸分子将解离为单链状态;随后,在步骤(d)中,互补的核酸分子(例如,正向引物与靶核酸或反向引物的延伸产物,反向引物与靶核酸或正向引物的延伸产物,第一通用引物与含有第一通用序列互补序列的扩增产物,第二通用引物与含有第二通用序列互补序列的扩增产物)将退火或杂交在一起,形成双链体;随后,在步骤(e)中,核酸聚合酶(特别是模板依赖性核酸聚合酶)将延伸与互补序列杂交的正向/反向引物和第一/第二通用引物。由此,通过步骤(c)-(e)的循环,可实现靶核酸序列的扩增(特别是如上文所详细说明的,不对称扩增),从而完成本发明方法的步骤(1)-(2)。
73.步骤(c)的温育时间和温度可以由本领域技术人员常规地确定。在某些优选的实施方案中,在步骤(c)中,在80-105℃(例如,80-85℃,85-90℃,90-95℃,91℃,92℃,93℃,94℃,95℃,96℃,97℃,98℃,99℃,100℃,101℃,102℃,103℃,104℃,或105℃)的温度下温育步骤(b)的产物,从而使核酸变性。在某些优选的实施方案中,在步骤(c)中,温育步骤(b)的产物10s-5min,例如10-20s,20-40s,40-60s,1-2min,或2-5min。
74.步骤(d)的温育时间和温度可以由本领域技术人员常规地确定。在某些优选的实施方案中,在步骤(d)中,在35-70℃(例如,35-40℃,40-45℃,45-50℃,50-55℃,55-60℃,60-65℃,或65-70℃)的温度下温育步骤(c)的产物,从而允许核酸退火或杂交。在某些优选的实施方案中,在步骤(d)中,温育步骤(c)的产物10s-5min,例如10-20s,20-40s,40-60s,1-2min,或2-5min。
75.步骤(e)的温育时间和温度可以由本领域技术人员常规地确定。在某些优选的实施方案中,在步骤(e)中,在35-85℃(例如,35-40℃,40-45℃,45-50℃,50-55℃,55-60℃,60-65℃,65-70℃,70-75℃,75-80℃,80-85℃)的温度下温育步骤(d)的产物,从而允许核酸延伸。在某些优选的实施方案中,在步骤(e)中,温育步骤(d)的产物10s-30min,例如10-20s,20-40s,40-60s,1-2min,2-5min,5-10min,10-20min或20-30min。
76.在某些实施方案中,可在不同的温度下进行步骤(d)和(e),即在不同的温度下进行核酸的退火和延伸。在某些实施方案中,可在相同的温度下进行步骤(d)和(e),即在相同的温度下进行核酸的退火和延伸。在此情况下,可将步骤(d)和(e)合并为一个步骤。
77.在本发明的方法中,可重复步骤(c)-(e)至少一次,例如至少2次,至少5次,至少10次,至少20次,至少30次,至少40次,或至少50次。然而,易于理解的是,当重复步骤(c)-(e)一次或多次时,每一个循环的步骤(c)-(e)所使用的条件不必是相同的。例如,可使用一种条件来进行前一部分循环(例如前5个,前10个,前20个循环)的步骤(c)-(e),随后使用另一种条件来进行剩余循环的步骤(c)-(e)。
78.本发明的方法能够对靶核酸进行多重、不对称扩增,产生大量单链核酸产物。因此,在某些情况下,本发明方法是特别有利的。例如,本发明方法的扩增产物可以用于测序,用于基因芯片检测,或用于熔解曲线分析。因此,在某些优选的实施方案中,本发明的方法还包括步骤:(3)对步骤(2)的产物进行测序。在某些优选的实施方案中,本发明的方法还包括步骤:(3)使用基因芯片对步骤(2)的产物进行检测。在某些优选的实施方案中,本发明的方法还包括步骤:(3)对步骤(2)的产物进行熔解曲线分析。
79.检测方法
80.在一个方面,本申请提供了一种检测样品中的一种或多种靶核酸的方法,其包括,(i)使用本发明的方法扩增样品中的一种或多种靶核酸;(ii)对步骤(i)的产物进行熔解曲线分析。
81.在某些优选的实施方案中,本发明的方法包括以下步骤:
82.(1)提供含有一种或多种靶核酸的样品;和,提供第一通用引物和第二通用引物,以及,针对每一种待扩增的靶核酸,提供一个靶特异性引物对和一个检测探针;其中,
83.所述第一通用引物包含第一通用序列;
84.所述第二通用引物包含第二通用序列,所述第二通用序列包含第一通用序列且在第一通用序列的3'端额外包含至少一个核苷酸;
85.所述靶特异性引物对能够扩增所述靶核酸,并且包含一个正向引物和一个反向引物,其中,所述正向引物包含第一通用序列和特异于所述靶核酸的正向核苷酸序列,且所述正向核苷酸序列位于第一通用序列的3'端;所述反向引物包含第二通用序列和特异于所述靶核酸的反向核苷酸序列,且所述反向核苷酸序列位于第二通用序列的3'端;并且,第二通用序列不能与所述正向引物的互补序列完全互补;
86.所述检测探针包含特异于所述靶核酸的探针核苷酸序列,并且标记有报告基团和淬灭基团,其中,所述报告基团能够发出信号,并且,所述淬灭基团能够吸收或淬灭所述报告基团发出的信号;并且,所述检测探针在与其互补序列杂交的情况下发出的信号不同于在未与其互补序列杂交的情况下发出的信号;
87.(2)在允许核酸扩增的条件下,使用所述第一通用引物、所述第二通用引物以及所述靶特异性引物对,通过pcr反应扩增样品中的靶核酸;
88.(3)使用所述检测探针对步骤(2)的产物进行熔解曲线分析;并根据熔解曲线分析的结果,确定所述靶核酸是否存在于样品中。
89.在本发明的某些实施方案中,样品、靶核酸、第一通用引物、第二通用引物和/或靶特异性引物对如上文所定义。
90.在本发明的某些实施方案中,在步骤(2)中,将所述样品与所述第一通用引物、所述第二通用引物和所述靶特异性引物对,以及核酸聚合酶混合,并进行pcr反应。然后,在pcr反应结束后,将检测探针加入到步骤(2)的产物中,并进行熔解曲线分析。
91.在本发明的某些实施方案中,在步骤(2)中,将所述样品与所述第一通用引物、所述第二通用引物、所述靶特异性引物对和所述检测探针,以及核酸聚合酶混合,并进行pcr反应。然后,在pcr反应结束后,进行熔解曲线分析。
92.在本发明的某些实施方案中,检测探针可以包含或者由天然存在的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸),经修饰的核苷酸,非天然的核苷酸(例如肽核酸(pna)或锁核酸),或其任何组合组成。在某些优选的实施方案中,检测探针包含或者由天然的核苷酸(例如脱氧核糖核苷酸或核糖核苷酸)组成。在某些优选的实施方案中,检测探针包含经修饰的核苷酸,例如经修饰的脱氧核糖核苷酸或核糖核苷酸,例如5-甲基胞嘧啶或5-羟甲基胞嘧啶。在某些优选的实施方案中,检测探针包含非天然的核苷酸,例如脱氧次黄嘌呤,肌苷,1-(2'-脱氧-β-d-呋喃核糖基)-3-硝基吡咯,5-硝基吲哚或锁核酸(lna)。
93.在本发明的方法中,检测探针不受其长度的限制。例如,检测探针的长度可以为
15-1000nt,例如15-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt,80-90nt,90-100nt,100-200nt,200-300nt,300-400nt,400-500nt,500-600nt,600-700nt,700-800nt,800-900nt,900-1000nt。
94.在某些优选的实施方案中,检测探针具有3'-oh末端。在某些优选的实施方案中,检测探针的3'-末端是封闭的,以抑制其延伸。可通过各种方法来封闭核酸(例如检测探针)的3'-末端。例如,可通过对检测探针的最后一个核苷酸的3'-oh进行修饰,以封闭检测探针的3'-末端。在某些实施方案中,可通过在检测探针的最后一个核苷酸的3'-oh上添加化学部分(例如,生物素或烷基),从而封闭检测探针的3'-末端。在某些实施方案中,可通过将检测探针的最后一个核苷酸的3'-oh去除,或者将所述最后一个核苷酸替换为双脱氧核苷酸,从而封闭检测探针的3'-末端。
95.如上文所描述的,检测探针标记有报告基团和淬灭基团,其中,所述报告基团能够发出信号,并且,所述淬灭基团能够吸收或淬灭所述报告基团发出的信号;并且,所述检测探针在与其互补序列杂交的情况下发出的信号不同于在未与其互补序列杂交的情况下发出的信号。
96.在某些优选的实施方案中,所述检测探针为自淬灭探针。在此类实施方案中,当检测探针未与其他序列杂交时,淬灭基团位于能够吸收或淬灭报告基团的信号的位置(例如,淬灭基团位于报告基团的邻近),从而吸收或淬灭报告基团发出的信号。在这种情况下,所述检测探针不发出信号。进一步,当所述检测探针与其互补序列杂交时,淬灭基团位于不能吸收或淬灭报告基团的信号的位置(例如,淬灭基团位于远离报告基团的位置),从而无法吸收或淬灭报告基团发出的信号。在这种情况下,所述检测探针发出信号。
97.此类自淬灭检测探针的设计在本领域技术人员的能力范围之内。例如,可在所述检测探针的5'末端标记报告基团而在3'末端标记淬灭基团,或可在所述检测探针的3'末端标记报告基团而在5'末端标记淬灭基团。由此,当所述检测探针单独存在时,所述报告基团与所述淬灭基团彼此接近并相互作用,使得所述报告基团发出的信号被所述淬灭基团吸收,从而使得所述检测探针不发出信号;而当所述检测探针与其互补序列杂交时,所述报告基团与所述淬灭基团相互分离,使得所述报告基团发出的信号不能被所述淬灭基团吸收,从而使得所述检测探针发出信号。
98.然而,应当理解的是,报告基团和淬灭基团并非必须标记在检测探针的末端。报告基团和/或淬灭基团也可以标记在检测探针的内部,只要所述检测探针在与其互补序列杂交的情况下发出的信号不同于在未与其互补序列杂交的情况下发出的信号。例如,可将报告基团标记在检测探针的上游(或下游),而将淬灭基团标记在检测探针的下游(或上游),并且二者相距足够的距离(例如相距10-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt,或更长的距离)。由此,当所述检测探针单独存在时,由于探针分子的自由卷曲或者探针的二级结构(例如发夹结构)的形成,所述报告基团与所述淬灭基团彼此接近并相互作用,使得所述报告基团发出的信号被所述淬灭基团吸收,从而使得所述检测探针不发出信号;并且,当所述检测探针与其互补序列杂交时,所述报告基团与所述淬灭基团相互分离足够的距离,使得所述报告基团发出的信号不能被所述淬灭基团吸收,从而使得所述检测探针发出信号。在某些优选的实施方案中,报告基团和淬灭基团相距10-80nt或更长的距离,例如10-20nt,20-30nt,30-40nt,40-50nt,50-60nt,60-70nt,70-80nt。在某些优选的
chemicals(molecular probes,eugene,1992);pringsheim,fluorescence and phosphorescence(interscience publishers,new york,1949);haugland,r.p.,handbook offluorescent probes and research chemicals,6th edition(molecular probes,eugene,oreg.,1996);美国专利3,996,345和4,351,760。
102.在某些优选的实施方案中,所述报告基团为荧光基团。在此类实施方案中,报告基团发出的信号即为荧光,并且,淬灭基团为能够吸收/淬灭所述荧光的分子或基团(例如,能够吸收所述荧光的另一荧光分子,或者能够淬灭所述荧光的淬灭剂)。在某些优选的实施方案中,所述荧光基团包括但不限于各种荧光分子,例如alex-350,fam,vic,tet,calgold 540,joe,hex,cal fluor orange 560,tamra,cal fluor red 590,rox,cal fluor red 610,texas red,cal fluor red 635,quasar 670,cy3,cy5,cy5.5,quasar 705等。在某些优选的实施方案中,所述淬灭基团包括但不限于各种淬灭剂,例如dabcyl、bhq(例如bhq-1或者bhq-2)、eclipse、和/或tamra等。
103.在本发明的方法中,还可以对检测探针进行修饰,例如使其具有抵抗核酸酶活性(例如5'核酸酶活性,例如5'至3'核酸外切酶活性)的抗性。例如,可在检测探针的主链中引入抵抗核酸酶活性的修饰,例如硫代磷酸酯键,烷基磷酸三酯键,芳基磷酸三酯键,烷基膦酸酯键,芳基膦酸酯键,氢化磷酸酯键,烷基氨基磷酸酯键,芳基氨基磷酸酯键,2'-o-氨基丙基修饰,2'-o-烷基修饰,2'-o-烯丙基修饰,2'-o-丁基修饰,和1-(4'-硫代-pd-呋喃核糖基)修饰。
104.在本发明的方法中,检测探针可以是线性的,或者可具有发夹结构。在某些优选的实施方案中,所述检测探针是线性的。在某些优选的实施方案中,所述检测探针具有发夹结构。发夹结构可以是天然的,也可以是人工引入的。此外,可使用本领域中的常规方法来构建具有发夹结构的检测探针。例如,可通过在检测探针的2个末端(5'端和3'端)添加互补的2段寡核苷酸序列,从而使得检测探针可形成发夹结构。在此类实施方案中,互补的2段寡核苷酸序列构成发夹结构的臂(茎)。发夹结构的臂可具有任何期望的长度,例如臂的长度可以是2-15nt,例如3-7nt,4-9nt,5-10nt,6-12nt。
105.在某些实施方案中,可对步骤(2)的产物进行逐渐的升温并实时监测检测探针上的报告基团发出的信号,从而获得步骤(2)的产物的信号强度随着温度变化而变化的曲线。例如,可将步骤(2)的产物从45℃或更低的温度(例如,不超过45℃,不超过40℃,不超过35℃,不超过30℃,不超过25℃)逐渐升温至75℃或更高的温度(例如,至少75℃,至少80℃,至少85℃,至少90℃,至少95℃),并实时监测检测探针上的报告基团发出的信号,从而获得所述报告基团的信号强度随着温度变化而变化的曲线。升温的速率可以由本领域技术人员常规地确定。例如,升温的速率可以为:每步骤升温0.01-1℃(例如0.01-0.05℃、0.05-0.1℃、0.1-0.5℃、0.5-1℃、0.04-0.4℃,例如0.01℃、0.02℃、0.03℃、0.04℃、0.05℃、0.06℃、0.07℃、0.08℃、0.09℃、0.1℃、0.2℃、0.3℃、0.4℃、0.5℃、0.6℃、0.7℃、0.8℃、0.9℃或1.0℃),并且每步骤维持0.5-15s(例如0.5-1s,1-2s,2-3s,3-4s,4-5s,5-10s,10-15s);或者每秒升温0.01-1℃(例如0.01-0.05℃、0.05-0.1℃、0.1-0.5℃、0.5-1℃、0.04-0.4℃,例如0.01℃、0.02℃、0.03℃、0.04℃、0.05℃、0.06℃、0.07℃、0.08℃、0.09℃、0.1℃、0.2℃、0.3℃、0.4℃、0.5℃、0.6℃、0.7℃、0.8℃、0.9℃或1.0℃)。
106.在某些实施方案中,可对步骤(2)的产物进行逐渐的降温并实时监测检测探针上
的报告基团发出的信号,从而获得步骤(2)的产物的信号强度随着温度变化而变化的曲线。例如,可将步骤(2)的产物从75℃或更高的温度(例如,至少75℃,至少80℃,至少85℃,至少90℃,至少95℃)逐渐降温至45℃或更低的温度(例如,不超过45℃,不超过40℃,不超过35℃,不超过30℃,不超过25℃),并实时监测检测探针上的报告基团发出的信号,从而获得所述报告基团的信号强度随着温度变化而变化的曲线。降温的速率可以由本领域技术人员常规地确定。例如,降温的速率可以为:每步骤降温0.01-1℃(例如0.01-0.05℃、0.05-0.1℃、0.1-0.5℃、0.5-1℃、0.04-0.4℃,例如0.01℃、0.02℃、0.03℃、0.04℃、0.05℃、0.06℃、0.07℃、0.08℃、0.09℃、0.1℃、0.2℃、0.3℃、0.4℃、0.5℃、0.6℃、0.7℃、0.8℃、0.9℃或1.0℃),并且每步骤维持0.5-15s(例如0.5-1s,1-2s,2-3s,3-4s,4-5s,5-10s,10-15s);或者每秒降温0.01-1℃(例如0.01-0.05℃、0.05-0.1℃、0.1-0.5℃、0.5-1℃、0.04-0.4℃,例如0.01℃、0.02℃、0.03℃、0.04℃、0.05℃、0.06℃、0.07℃、0.08℃、0.09℃、0.1℃、0.2℃、0.3℃、0.4℃、0.5℃、0.6℃、0.7℃、0.8℃、0.9℃或1.0℃)。
107.随后,可对获得的曲线进行求导,从而获得步骤(2)的产物的熔解曲线。根据熔解曲线中的熔解峰(熔点),可确定对应于该熔解峰(熔点)的靶核酸的存在。
108.在本发明的方法中,所使用的检测探针各自独立地可以使用相同或不同的报告基团。在某些优选的实施方案中,所使用的检测探针具有相同的报告基团。在此情况下,可对步骤(2)的产物进行熔解曲线分析,并根据熔解曲线中的熔解峰(熔点)来确定某一靶核酸的存在。在某些优选的实施方案中,所使用的检测探针具有不同的报告基团。在此情况下,当对步骤(2)的产物进行熔解曲线分析时,可分别实时监测每一种报告基团的信号,由此获得各自与一种报告基团的信号对应的多条熔解曲线。随后,可根据报告基团的信号种类以及熔解曲线中的熔解峰(熔点)来确定某一靶核酸的存在。由此,本发明的方法可实现对一种或多种(例如2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多种)靶核酸序列的同时检测(多重检测)。
109.不拘于理论限制,针对同一报告基团(同一熔解曲线),熔解曲线分析的分辨率或精度可达到0.5℃或更高。换言之,熔解曲线分析能够区分同一熔解曲线中熔点相差仅0.5℃或更低(例如0.1℃、0.2℃、0.3℃、0.4℃、0.5℃)的两个熔解峰。因此,在本发明方法的某些实施方案中,在使用相同报告基团的情况下,各种靶核酸与其检测探针所形成的各种双链体之间的熔点差异可以为至少0.5℃,从而不同的双链体(由此,不同的靶核酸)可通过熔解曲线分析来区分和辨别。然而,出于便于区分和辨别的目的,两个双链体的更大的熔点差异在某些情况下是优选的。因此,在本发明方法的某些实施方案中,两个双链体之间的熔点差异可以为任何期望的值(例如至少0.5℃,至少1℃,至少2℃,至少3℃,至少4℃,至少5℃,至少8℃,至少10℃,至少15℃,或至少20℃),只要所述熔点差异能够通过熔解曲线分析来区分和辨别即可。
110.在某些优选的实施方案中,本发明方法的步骤(1)-(3)可通过包含下述步骤(a)-(g)的方案来进行:
111.(a)提供含有一种或多种靶核酸的样品;和,提供第一通用引物和第二通用引物,以及,针对每一种待扩增的靶核酸,提供一个靶特异性引物对和一个检测探针;其中,所述第一通用引物、第二通用引物、靶特异性引物对和检测探针如上文所定义;
112.(b)将所述样品与所述第一通用引物、第二通用引物、靶特异性引物对和检测探
针,以及核酸聚合酶(例如,模板依赖性核酸聚合酶;例如dna聚合酶,特别是热稳定的dna聚合酶)混合;
113.(c)在允许核酸变性的条件下,温育前一步骤的产物;
114.(d)在允许核酸退火或杂交的条件下,温育前一步骤的产物;
115.(e)在允许核酸延伸的条件下,温育前一步骤的产物;
116.(f)任选地,重复步骤(c)-(e)一次或多次;和
117.(g)对前一步骤的产物进行熔解曲线分析。
118.关于步骤(a)-(g),其已详细描述于上文中。
119.引物组和试剂盒
120.在一个方面,本发明提供了一种引物组,其包括:第一通用引物和第二通用引物,以及,一个或多个靶特异性引物对;其中,
121.所述第一通用引物包含第一通用序列;
122.所述第二通用引物包含第二通用序列,所述第二通用序列包含第一通用序列且在第一通用序列的3'端额外包含至少一个核苷酸;
123.每个靶特异性引物对各自能够扩增一个靶核酸,并且包含一个正向引物和一个反向引物,其中,所述正向引物包含第一通用序列和特异于所述靶核酸的正向核苷酸序列,且所述正向核苷酸序列位于第一通用序列的3'端;所述反向引物包含第二通用序列和特异于所述靶核酸的反向核苷酸序列,且所述反向核苷酸序列位于第二通用序列的3'端;并且,第二通用序列不能与所述正向引物的互补序列完全互补。
124.在某些优选的实施方案中,所述引物组包含1-5个,5-10个,10-15个,15-20个,20-50个或更多个靶特异性引物对,例如,至少2个,至少3个,至少4个,至少5个,至少8个,至少10个,至少12个,至少15个,至少18个,至少20个,至少25个,至少30个,至少40个,至少50个,或更多个靶特异性引物对。
125.易于理解的是,此类引物组可用于实施上文所详细描述的本发明方法。因此,上文针对第一通用引物、第二通用引物、靶特异性引物对、靶核酸、样品所详细描述的各种技术特征同样可应用于本申请中涉及引物组的技术方案。因此,在某些优选的实施方案中,所述引物组包含如上文所定义的第一通用引物、第二通用引物和/或靶特异性引物对。
126.在某些优选的实施方案中,所述引物组可用于扩增样品中的1种靶核酸,其包括:第一通用引物和第二通用引物,以及,包含第一正向引物和第一反向引物的第一靶特异性引物对;其中,
127.所述第一通用引物包含第一通用序列;
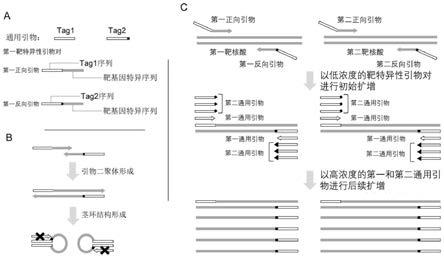
128.所述第二通用引物包含第二通用序列,所述第二通用序列包含第一通用序列且在第一通用序列的3'端额外包含至少一个核苷酸(例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸);
129.第一正向引物包含第一通用序列和特异于第一靶核酸的第一正向核苷酸序列,且第一正向核苷酸序列位于第一通用序列的3'端;
130.第一反向引物包含第二通用序列和特异于第一靶核酸的第一反向核苷酸序列,且第一反向核苷酸序列位于第二通用序列的3'端;并且,
131.第一正向引物和第一反向引物能够特异性扩增第一靶核酸;并且,
132.第二通用序列不能与第一正向引物的互补序列完全互补。
133.在某些优选的实施方案中,所述引物组可用于扩增样品中的2种靶核酸,其包括:第一通用引物和第二通用引物,以及,第一靶特异性引物对和第二靶特异性引物对;其中,第一通用引物和第二通用引物如上文所定义,并且,第一靶特异性引物对包含能够扩增第一靶核酸的第一正向引物和第一反向引物,第二靶特异性引物对包含能够扩增第二靶核酸的第二正向引物和第二反向引物;其中,第一正向引物、第一反向引物、第二正向引物和第二反向引物如上文所定义。类似地,在某些优选的实施方案中,本发明的引物组可用于扩增样品中的3种或更多种靶核酸,其包括:第一通用引物,第二通用引物,以及能够扩增所述3种或更多种靶核酸的3种或更多种靶特异性引物对。
134.另外,为了方便,可以将本发明的引物组与实施本发明方法(扩增方法或检测方法)所需的一种或多种试剂组合,制备成试剂盒。易于理解的是,此类试剂盒可用于实施上文所详细描述的本发明方法。因此,上文针对各种成分所详细描述的各种技术特征同样可应用于试剂盒中的各种成分。并且,此类试剂盒还可包含实施本发明方法所需的其他试剂。
135.因此,在另一个方面,本申请提供了一种试剂盒,其包含如上所述的引物组,以及,选自下列的一种或多种组分:核酸聚合酶,用于进行核酸扩增的试剂,用于进行测序的试剂,用于进行基因芯片检测的试剂,用于进行熔解曲线分析的试剂,或其任何组合。
136.在某些优选的实施方案中,所述核酸聚合酶是模板依赖性核酸聚合酶,例如dna聚合酶,特别是热稳定的dna聚合酶。在某些优选的实施方案中,所述核酸聚合酶如上文所定义。
137.用于进行核酸扩增的试剂可由本领域技术人员常规地确定,并且包括但不限于,酶(例如核酸聚合酶)的工作缓冲液、dntps(标记或未标记的)、水、包含离子(例如mg
2+
)的溶液、单链dna结合蛋白(single strand dna-binding protein,ssb)、或其任何组合。
138.用于进行测序的试剂可由本领域技术人员常规地确定,并且包括但不限于,酶(例如核酸聚合酶)的工作缓冲液、dntps(标记或未标记的)、ddntps(标记或未标记的)、水、包含离子(例如mg
2+
)的溶液、单链dna结合蛋白(single strand dna-binding protein,ssb)、连接酶、核酸接头、测序引物或其任何组合。
139.用于进行基因芯片检测的试剂可由本领域技术人员常规地确定,并且包括但不限于,酶(例如核酸聚合酶)的工作缓冲液、dntps(标记或未标记的)、水、杂交缓冲液、洗涤缓冲液、标记试剂或其任何组合。
140.用于进行熔解曲线分析的试剂可由本领域技术人员常规地确定,并且包括但不限于,检测探针。在某些优选的实施方案中,所述检测探针是自淬灭探针,例如自淬灭荧光探针。在某些优选的实施方案中,所述检测探针如上文所定义。
141.本领域技术人员基于本申请所详细描述的原理,可对本发明技术方案的各种技术特征进行修饰、替换或组合,而不背离本发明的精神和范围。所有此类技术方案以及其变形都涵盖在本申请的权利要求书或其等同物的范围内。
142.发明的有益效果
143.与现有技术相比,本发明的技术方案具有以下有益效果:
144.(1)相比于常规的hand系统和使用常规hand系统的扩增方法而言,本发明的技术方案能够实现对多种靶核酸的同时、不对称扩增(多重、不对称扩增)。
145.(2)相比于常规的多重、不对称pcr扩增方法而言,本发明的技术方案能够有效抑制引物二聚体的非特异性扩增,显著提高了扩增的特异性和检测的灵敏度。
146.因此,本发明开发了一种新的能够同时、不对称扩增多个靶核酸的方法,其能够同时实现有效的多重pcr扩增和不对称pcr扩增,满足临床上对多个靶核酸同时进行扩增和检测的要求。
147.下面将结合附图和实施例对本发明的实施方案进行详细描述,但是本领域技术人员将理解,下列附图和实施例仅用于说明本发明,而不是对本发明的范围的限定。根据附图和优选实施方案的下列详细描述,本发明的各种目的和有利方面对于本领域技术人员来说将变得显然。
附图说明
148.图1示意性地描述了本发明方法的示例性实施方案,以阐释本发明方法的基本原理。
149.图1a示意性地描述了该实施方案中所使用的引物组,其包括:第一通用引物和第二通用引物,以及,包含第一正向引物和第一反向引物的第一靶特异性引物对;其中,
150.第一通用引物包含第一通用序列(tag1);
151.第二通用引物包含第二通用序列(tag2),所述第二通用序列包含第一通用序列且在第一通用序列的3'端额外包含至少一个核苷酸(例如1-5个,5-10个,10-15个,15-20个或更多个核苷酸);
152.第一正向引物包含第一通用序列和特异于第一靶核酸的第一正向核苷酸序列,且第一正向核苷酸序列位于第一通用序列的3'端;
153.第一反向引物包含第二通用序列和特异于第一靶核酸的第一反向核苷酸序列,且第一反向核苷酸序列位于第二通用序列的3
′
端;并且,
154.第一正向引物和第一反向引物能够特异性扩增第一靶核酸;并且,
155.第二通用序列不能与第一正向引物的互补序列完全互补。
156.图1b示意性地描述了使用图1a的引物组进行扩增时,引物二聚体的非特异性扩增被抑制的原理,其中,因第一正向引物和第一反向引物的非特异性扩增而形成的引物二聚体在变性后将产生其5'端和3'端包含彼此互补的反向序列的单链核酸,该单链核酸自身在退火阶段将形成锅柄结构,阻止第一通用引物和第二通用引物对该单链核酸的退火和延伸,从而抑制引物二聚体的进一步扩增。
157.图1c示意性地描述了使用图1a的引物组进行多重、不对称扩增的原理。在该实施方案中,首先,由浓度低的第一靶特异性引物对启动pcr扩增,产生初始扩增产物,其包含分别与第一正向引物/第一通用引物和第一反向引物/第二通用引物互补的两条核酸链(核酸链a和核酸链b);随后,由浓度高的第一通用引物和第二通用引物对所述初始扩增产物进行后续的pcr扩增。
158.由于第一反向引物/第二通用引物包含第一通用序列,因此,第一通用引物不仅能够退火至核酸链a(与第一正向引物/第一通用引物互补的核酸链)并合成其互补链,而且能够退火至核酸链b(与第一反向引物/第二通用引物互补的核酸链)并合成其互补链。也即,第一通用引物可以同时扩增核酸链a和核酸链b。
159.第二通用引物在第一通用序列的3'端包含额外的核苷酸,因此,其与核酸链a(与第一正向引物/第一通用引物互补的核酸链)在3'端是不匹配的(即,在3'端不能完全互补)。由此,在扩增过程中,第二通用引物将优先退火至核酸链b(与第一反向引物/第二通用引物互补的核酸链)并合成其互补链,而基本上不能延伸合成核酸链a(与第一正向引物/第一通用引物互补的核酸链)的互补链。
160.因此,随着pcr扩增的进行,核酸链a的互补链(核酸链b)的合成效率将显著低于核酸链b的互补链(核酸链a),导致核酸链b的互补链(核酸链a)被大量合成和扩增,而核酸链a的互补链(核酸链b)的合成和扩增受到抑制,从而产生大量目标单链产物(核酸链a,其含有与第一正向引物/第一通用引物互补的序列以及第一反向引物/第二通用引物的序列),实现不对称扩增。此外,为进一步增强扩增的不对称性,还可以调整第一通用引物和第二通用引物的比例,使得第一通用引物的浓度低于第二通用引物,以更好地富集目标单链产物。
161.进一步,还可以将如上所定义的第一通用引物和第二通用引物与至少两个或更多个如上所定义的靶特异性引物对组合使用,其中,每一个靶特异性引物对各自包含一个正向引物和一个反向引物,且能够特异性扩增一种靶核酸,其中,所述正向引物包含第一通用序列和特异于所述靶核酸的正向核苷酸序列,所述反向引物包含第二通用序列和特异于所述靶核酸的反向核苷酸序列,由此,本发明的实施方案(引物组)可以用于实现对至少两个或更多个靶核酸的多重、不对称扩增。
162.在图1c所示的示例性实施方案中,将如上所定义的第一通用引物和第二通用引物与两个靶特异性引物对组合使用,其中,第一靶特异性引物对包含第一正向引物和第一反向引物,且能够特异性扩增第一靶核酸,并且,第一正向引物包含第一通用序列和特异于第一靶核酸的第一正向核苷酸序列,第一反向引物包含第二通用序列和特异于第一靶核酸的第一反向核苷酸序列;第二靶特异性引物对包含第二正向引物和第二反向引物,且能够特异性扩增第二靶核酸,并且,第二正向引物包含第一通用序列和特异于第二靶核酸的第二正向核苷酸序列,第二反向引物包含第二通用序列和特异于第二靶核酸的第二反向核苷酸序列。由此,该引物组可以用于实现对第一和第二靶核酸的同时、不对称扩增。
163.图2显示了实施例1中使用hand系统、普通不对称pcr系统以及本发明系统进行实时pcr扩增的结果;其中,黑色和灰色虚线分别代表使用hand系统扩增人类基因组dna和阴性对照的扩增曲线;黑色和灰色点线分别代表使用普通不对称pcr系统扩增人类基因组dna和阴性对照的扩增曲线;黑色和灰色实线分别代表使用本发明系统扩增人类基因组dna和阴性对照的扩增曲线。
164.图3显示了实施例1中使用hand系统、普通不对称pcr系统以及本发明系统扩增后进行熔解曲线分析的结果;其中,黑色和灰色虚线分别代表使用hand系统扩增人类基因组dna和阴性对照后进行熔解曲线分析的结果;黑色和灰色点线分别代表使用普通不对称pcr系统扩增人类基因组dna和阴性对照后进行熔解曲线分析的结果;黑色和灰色实线分别代表使用本发明系统扩增人类基因组dna和阴性对照后进行熔解曲线分析的结果。
165.图4显示了实施例1中使用hand系统、普通不对称pcr系统以及本发明系统获得的扩增产物的琼脂糖凝胶电泳结果;其中,泳道m代表分子量标记;泳道1-3分别代表用hand系统(泳道1)、本发明系统(泳道2)和普通不对称pcr系统(泳道3)扩增人类基因组dna的产物;泳道4-6分别代表用hand系统、本发明系统和普通不对称pcr系统扩增阴性对照的产物。
166.图5显示了实施例2中使用具有不同比例的第一和第二通用引物的本发明系统扩增后进行熔解曲线分析的结果。
167.图6显示了实施例3中使用本发明系统扩增后进行熔解曲线分析的结果,其中,黑色实线代表使用本发明系统扩增样本1后进行熔解曲线分析的结果;黑色虚线代表使用本发明系统扩增样本2后进行熔解曲线分析的结果;灰色实线代表使用本发明系统扩增阴性对照后进行熔解曲线分析的结果。
168.图7显示了实施例4中使用本发明系统扩增后进行熔解曲线分析的结果,其中,黑色实线(样本3)、黑色虚线(样本4)、黑色点线(样本5)、灰色虚线(样本6)、灰色点线(样本7)分别代表使用本发明系统扩增样本3-7后进行熔解曲线分析的结果;灰色实线代表使用本发明系统扩增阴性对照后进行熔解曲线分析的结果。
169.图8显示了实施例5中使用本发明系统扩增后进行熔解曲线分析的结果,其中,黑色点线、黑色虚线、灰色点线、灰色虚线、黑色实线、灰色实线分别代表,对基因组dna浓度为10ng/μl、1ng/μl、0.1ng/μl、0.05ng/μl、0.02ng/μl或0.01ng/μl的样品进行扩增后进行熔解曲线分析的结果。
170.图9显示了实施例5中使用普通多重不对称pcr系统扩增后进行熔解曲线分析的结果,其中,黑色点线、黑色虚线、灰色点线、灰色虚线、黑色实线、灰色实线分别代表,对基因组dna浓度为10ng/μl、1ng/μl、0.1ng/μl、0.05ng/μl、0.02ng/μl或0.01ng/μl的样品进行扩增后进行熔解曲线分析的结果。
具体实施方式
171.现参照下列意在举例说明本发明(而非限定本发明)的实施例来描述本发明。应当理解的是,这些实施例只是用于说明本发明的原理和技术效果,而并不是表示本发明的所有可能性。本发明并不局限于这些实施例中提到的材料、反应条件或参数。本领域技术人员可以根据本发明的原理,利用其它类似的材料或反应条件来实施其他技术方案。此类技术方案没有脱离本发明描述的基本原理和概念,并且涵盖在本发明的范围内。
172.实施例1
173.在本实施例中,以人15号染色体上的涵盖基因多态性位点rs8027171的dna片段为待扩增的靶核酸,考察了hand系统、普通不对称pcr系统以及本发明系统(引物组)产生单链核酸产物的情况。本实施例所使用的引物和探针的序列如表1所示。本实施例采用的仪器为slan 96实时荧光pcr仪(厦门致善生物科技股份有限公司,厦门)。
174.简言之,在本实施例中使用25μl的pcr反应体系来进行pcr扩增和熔解曲线分析,所述pcr反应体系包括:1
×
taq pcr buffer(takara,北京),5.0mm mgcl2,0.2mm dntps,1u taq聚合酶(takara,北京),0.3μm rs8027171-p探针,5μl人类基因组dna(rs8027171的基因型为g/a杂合)或阴性对照(水),以及引物;其中,
175.(1)hand系统所使用的引物为0.02μm rs8027171-f,0.02μm rs8027171-r,0.66μm tag 1引物;
176.(2)普通不对称pcr系统所使用的引物为0.06μm rs8027171-f,0.6μm rs8027171-r;和
177.(3)本发明系统所使用的引物为0.02μm rs8027171-f,0.02μm rs8027171-r,0.06
μm tag 1引物,0.6μm tag 2引物。
178.pcr扩增程序为:95℃预变性5min;10个循环的(95℃变性15s,65℃-56℃退火15s(每个循环下降1℃),76℃延伸20s);50个循环的(95℃变性15s,55℃退火15s,76℃延伸20s);并且在退火阶段采集cy5通道的荧光信号。在pcr扩增结束后,进行熔解曲线分析,程序为:95℃变性1min;37℃保温3min;随后,按0.04℃/s的升温速率从40℃递增至85℃,并采集cy5通道的荧光信号。最后,各pcr产物用2%琼脂糖凝胶电泳进行分析。实验结果如图2-4所示。
179.表1:实施例1所使用的引物和探针的序列
[0180][0181]
图2显示了实施例1中使用hand系统、普通不对称pcr系统以及本发明系统进行实时pcr扩增的结果;其中,黑色和灰色虚线分别代表使用hand系统扩增人类基因组dna和阴性对照的扩增曲线;黑色和灰色点线分别代表使用普通不对称pcr系统扩增人类基因组dna和阴性对照的扩增曲线;黑色和灰色实线分别代表使用本发明系统扩增人类基因组dna和阴性对照的扩增曲线。结果显示,3种系统均能够有效且特异性扩增人类基因组dna,产生对应的扩增信号(黑色虚线、黑色点线和黑色实线);并且各系统的阴性对照均无扩增信号产生(灰色虚线、灰色点线和灰色实线)。另外,还注意到,普通不对称pcr系统的扩增曲线(黑色点线)的ct值最小,这是由高浓度的靶特异性引物直接扩增靶核酸所导致的。
[0182]
图3显示了实施例1中使用hand系统、普通不对称pcr系统以及本发明系统扩增后进行熔解曲线分析的结果;其中,黑色和灰色虚线分别代表使用hand系统扩增人类基因组dna和阴性对照后进行熔解曲线分析的结果;黑色和灰色点线分别代表使用普通不对称pcr系统扩增人类基因组dna和阴性对照后进行熔解曲线分析的结果;黑色和灰色实线分别代表使用本发明系统扩增人类基因组dna和阴性对照后进行熔解曲线分析的结果。
[0183]
图4显示了实施例1中使用hand系统、普通不对称pcr系统以及本发明系统获得的扩增产物的琼脂糖凝胶电泳结果;其中,泳道m代表分子量标记;泳道1-3分别代表用hand系统(泳道1)、本发明系统(泳道2)和普通不对称pcr系统(泳道3)扩增人类基因组dna的产物;泳道4-6分别代表用hand系统、本发明系统和普通不对称pcr系统扩增阴性对照的产物。
[0184]
图3-4的结果显示,当使用hand系统进行扩增时,扩增产物基本上为双链核酸,无法产生单链核酸产物(图4,泳道1);相应地,在熔解曲线分析过程中,探针无法与扩增产物中的互补链有效杂交,故不能产生有效的熔解峰(图3,黑色虚线)。因此,当使用hand系统进行扩增后,通常无法继续进行有效的探针熔解曲线分析。而当使用本发明系统和普通不对称pcr系统进行扩增时,均产生了大量的单链核酸产物(图4,泳道2和3);由此,在熔解曲线分析过程中,探针能够与扩增产物有效杂交,产生特异的熔解峰(图3,黑色实线和黑色点
线)。另外,图3-4的结果还显示,使用水为模板的阴性对照体系均不能产生有效的熔解峰(图3的灰色虚线、灰色点线和灰色实线),这是因为在pcr过程中无目的扩增产物产生(图4,泳道4-6)。此外,如图3所示,黑色点线和黑色实线在相同的位置处出现了两个熔解峰。这一结果表明,所检测的样品(靶核酸)为杂合型,其在扩增过程中产生了2种单链核酸产物。
[0185]
本实施例的结果表明,本发明的系统可用于实现靶核酸的不对称扩增,从而可与探针熔解曲线分析联合使用。
[0186]
实施例2
[0187]
在本实施例中,以人15号染色体上的涵盖基因多态性位点rs8027171的dna片段为待扩增的靶核酸,考察了第一和第二通用引物的比例对不对称扩增的影响。本实施例所使用的引物和探针的序列如表1所示。本实施例采用的仪器为slan 96实时荧光pcr仪。
[0188]
简言之,在本实施例中使用25μl的pcr反应体系来进行pcr扩增和熔解曲线分析,所述pcr反应体系包括:1
×
taq pcr buffer,5.0mm mgcl2,0.2mm dntps,1u taq dna聚合酶,5μl人类基因组dna(rs8027171的基因型为g/a杂合),0.02μm rs8027171-f,0.02μm rs8027171-r,0.3μm rs8027171-p探针,0.06μm tag 1引物,以及,如下用量的tag 2引物:0.06μm(tag 1/tag 2=1/1),0.24μm(tag 1/tag 2=1/4),0.48μm(tag 1/tag 2=1/8),0.72μm(tag 1/tag 2=1/12),0.96μm(tag 1/tag 2=1/16),或1.2μm(tag 1/tag 2=1/20)。
[0189]
pcr扩增程序为:95℃预变性5min;10个循环的(95℃变性15s,65℃-56℃退火15s(每个循环下降1℃),76℃延伸20s);50个循环的(95℃变性15s,55℃退火15s,76℃延伸20s)。在pcr扩增结束后,进行熔解曲线分析,程序为:95℃变性1min;37℃维持3min;随后,按0.04℃/s的升温速率从40℃递增至85℃,并采集cy5通道的荧光信号。实验结果如图5所示。
[0190]
图5显示了实施例2中使用具有不同比例的第一和第二通用引物的本发明系统扩增后进行熔解曲线分析的结果。图5的结果显示,在tag 1/tag2≤1/1的各种比例下,本发明的系统均可用于实现靶核酸的不对称扩增,产生单链核酸产物,从而,其扩增产物可用于进行有效的熔解曲线分析。图5的结果还显示,不同的tag 1/tag 2比例可能对熔解峰的高度有所影响。这是因为不同的tag 1/tag 2比例可能影响pcr反应的扩增效率,导致单链核酸产物的生成量有所不同。适合于本发明系统的最优的tag1/tag 2比例可根据实际情况,通过实验来调整和确定。
[0191]
实施例3
[0192]
在本实施例中,以基因多态性位点rs48189298和rs60871880的分型为例,说明本发明系统可以在单个反应管中实现双重、不对称扩增,并用于探针熔解曲线分析。本实施例所使用的引物和探针的序列如表2所示。本实施例采用的仪器为slan 96实时荧光pcr仪。
[0193]
简言之,在本实施例中使用25μl的pcr反应体系来进行pcr扩增和熔解曲线分析,所述pcr反应体系包括:1
×
taq pcr buffer,5.0mm mgcl2,0.2mm dntps,1u taq dna聚合酶,0.04μm rs48189298-f,0.04μm rs48189298-r,0.4μm rs48189298-p,0.06μm rs60871880-f,0.06μm rs60871880-r,0.4μm rs60871880-p,0.2μm tag 3引物,1.6μm tag 2引物,5μl人类基因组dna或阴性对照(水)。在本实施例中,检测了两个样本(样本1和2),其中,样本1的rs48189298和rs60871880位点的基因型经测序确定为a/a和g/g;样本2的
rs48189298和rs60871880位点的基因型经测序确定为a/t和g/a。
[0194]
pcr扩增程序为:95℃预变性5min;10个循环的(95℃变性15s,65℃-56℃退火15s(每个循环下降1℃),76℃延伸20s);50个循环的(95℃变性15s,55℃退火15s,76℃延伸20s)。在pcr扩增结束后,进行熔解曲线分析,程序为:95℃变性1min;37℃维持3min;随后,按0.04℃/s的升温速率从40℃递增至85℃,并采集rox通道的荧光信号。实验结果如图6所示。
[0195]
表2:实施例3所使用的引物和探针的序列
[0196][0197]
图6显示了实施例3中使用本发明系统扩增后进行熔解曲线分析的结果,其中,黑色实线代表使用本发明系统扩增样本1后进行熔解曲线分析的结果;黑色虚线代表使用本发明系统扩增样本2后进行熔解曲线分析的结果;灰色实线代表使用本发明系统扩增阴性对照后进行熔解曲线分析的结果。
[0198]
图6的结果显示,样本1(黑色实线)的基因多态性位点rs48189298和rs60871880的基因型分别为a/a和g/g;样本2(黑色虚线)的基因多态性位点rs48189298和rs60871880位点的基因型分别为a/t和g/a;阴性对照(灰色实线)无熔解峰;各样本的基因分型结果与测序获得的结果一致。这些结果表明,本发明系统能够在单个反应体系中同时对两个靶核酸进行不对称扩增,并分别产生足够的单链核酸产物,用于进行有效的、可靠的熔解曲线分析,进而实现对两个靶核酸的鉴定(例如,对两个基因多态性位点的基因分型)。因此,结合熔解曲线分析技术,本发明系统能够同时实现对两个靶核酸的检测和鉴定(例如基因分型)。
[0199]
实施例4
[0200]
在本实施例中,以13个基因多态性位点(即,rs4847034、rs2826949、rs8103778、rs1396009、rs1523537、rs1528460、rs7937238、rs2111980、rs7278737、rs591173、rs1358856、rs2730648和rs859400)的分型为例,说明本发明系统可以在单个反应管中实现13重、不对称扩增,并用于探针熔解曲线分析。本实施例所使用的引物和探针的序列如表3所示。本实施例采用的仪器为slan 96实时荧光pcr仪。
[0201]
简言之,在本实施例中使用25μl的pcr反应体系来进行pcr扩增和熔解曲线分析,所述pcr反应体系包括:1
×
taq pcr buffer,5.0mm mgcl2,0.2mm dntps,1u taq dna聚合酶,5μl人类基因组dna或阴性对照(水),以及,引物和探针。各引物和探针的使用浓度如表3所示。在本实施例中,共检测了5个样本(样本3-7;每个pcr反应体系检测一个样本)。
[0202]
pcr扩增程序为:95℃预变性5min;10个循环的(95℃变性15s,65℃-56℃退火15s
(每个循环下降1℃),76℃延伸20s);50个循环的(95℃变性15s,55℃退火15s,76℃延伸20s)。在pcr扩增结束后,进行熔解曲线分析,程序为:95℃变性1min;37℃维持3min;随后,按0.04℃/s的升温速率从40℃递增至85℃,并采集fam、hex、rox、cy5和quasar 705通道的荧光信号。实验结果如图7所示。
[0203]
表3:实施例4所使用的引物和探针的序列
[0204][0205][0206]
图7显示了实施例4中使用本发明系统扩增后进行熔解曲线分析的结果,其中,黑
色实线(样本3)、黑色虚线(样本4)、黑色点线(样本5)、灰色虚线(样本6)、灰色点线(样本7)分别代表使用本发明系统扩增样本3-7后进行熔解曲线分析的结果;灰色实线代表使用本发明系统扩增阴性对照后进行熔解曲线分析的结果。将图7的熔解曲线分析结果进一步概述于表4。
[0207]
表4:样本3-7的13个基因多态性位点的分型结果
[0208][0209][0210]
另外,还通过测序对样本3-7的13个基因多态性位点的基因分型结果进行了鉴定。结果显示,利用本发明系统获得的各样本的基因分型结果(图7和表4)与通过测序获得的结果完全一致。
[0211]
这些结果表明,本发明系统能够在单个反应体系中同时对样品中的13个靶核酸进行不对称扩增,并分别产生足够的单链核酸产物,用于进行有效的、可靠的熔解曲线分析,进而实现对多个靶核酸的鉴定(例如,对多个基因多态性位点的基因分型)。因此,结合熔解曲线分析技术,本发明系统能够同时实现对多个靶核酸的检测和鉴定(例如基因分型)。
[0212]
实施例5
[0213]
在本实施例中,以含有不同浓度的人类基因组dna的样品的基因分型为例,比较了本发明系统和普通多重不对称pcr系统的分析灵敏度。本实施例所使用的基因组dna的13个基因多态性位点为已知基因型,具体如下:rs4847034:a/g;rs2826949:t/t;rs8103778:c/c;rs1396009:a/g;rs1523537:t/c;rs1528460:t/t;rs7937238:t/c;rs2111980:a/g;rs7278737:t/g;rs591173:t/c;rs1358856:a/a;rs2730648:g/a;和rs859400:g/g。本实施例所使用的引物和探针的序列如表3所示。本实施例采用的仪器为slan 96实时荧光pcr仪。
[0214]
简言之,在本实施例中使用25μl的pcr反应体系来进行pcr扩增和熔解曲线分析,所述pcr反应体系包括:1
×
taq pcr buffer,7.0mm mgcl2,0.2mm dntps,1u taq dna聚合酶,5μl人类基因组dna(浓度分别为10ng/μl、1ng/μl、0.1ng/μl、0.05ng/μl、0.02ng/μl或0.01ng/μl),以及引物和探针。各引物和探针的使用浓度如表5所示。
[0215]
pcr扩增程序为:95℃预变性5min;10个循环的(95℃变性15s,65℃-56℃退火15s(每个循环下降1℃),76℃延伸20s);50个循环的(95℃变性15s,55℃退火15s,76℃延伸20s)。在pcr扩增结束后,进行熔解曲线分析,程序为:95℃变性1min;37℃维持3min;随后,按0.04℃/s的升温速率从40℃递增至85℃,并采集fam、hex、rox、cy5和quasar 705通道的
荧光信号。实验结果如图8和9所示。
[0216]
表5:实施例5所使用的引物和探针的浓度
[0217]
[0218]
图8显示了实施例5中使用本发明系统扩增后进行熔解曲线分析的结果,其中,黑色点线、黑色虚线、灰色点线、灰色虚线、黑色实线、灰色实线分别代表,对基因组dna浓度为10ng/μl、1ng/μl、0.1ng/μl、0.05ng/μl、0.02ng/μl或0.01ng/μl的样品进行扩增后进行熔解曲线分析的结果。
[0219]
图9显示了实施例5中使用普通多重不对称pcr系统扩增后进行熔解曲线分析的结果,其中,黑色点线、黑色虚线、灰色点线、灰色虚线、黑色实线、灰色实线分别代表,对基因组dna浓度为10ng/μl、1ng/μl、0.1ng/μl、0.05ng/μl、0.02ng/μl或0.01ng/μl的样品进行扩增后进行熔解曲线分析的结果。
[0220]
图8的结果显示,即使人类基因组dna的浓度低至0.05ng/μl(灰色虚线),本发明系统仍然可以稳定、准确地检测出所有13个基因多态性位点的基因型。相比之下,图9的结果显示,仅当人类基因组dna的浓度达到10ng/μl(黑色点线)时,普通多重不对称pcr系统才能检测出所有13个基因多态性位点的基因型;当人类基因组dna的浓度为1ng/μl(黑色虚线)时,部分基因多态性位点(例如位点rs8103778)的基因型已无法检测和辨别(无法产生可识别的熔解峰)。这可能是因为在普通多重不对称pcr的反应体系中,高浓度的多种引物和探针相互影响,导致部分基因多态性位点的不对称扩增已无法有效进行,无法产生足量的单链产物用于熔解曲线分析。
[0221]
上述结果表明,本发明系统的检测灵敏度显著高于普通多重不对称pcr系统。这主要是因为本发明系统采用了低浓度的靶特异性引物和高浓度的通用引物来进行扩增,有效降低了引物之间的干扰,减少了二聚体等非特异性扩增,使得反应体系中各个靶核酸的扩增能够达到平衡,从而提升了整个反应体系的检测灵敏度。
[0222]
尽管本发明的具体实施方式已经得到详细的描述,但本领域技术人员将理解:根据已经公开的所有教导,可以对细节进行各种修改和变动,并且这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1