一种毕赤酵母中IPTG诱导的tRNA元件及其构建方法和应用
一种毕赤酵母中iptg诱导的trna元件及其构建方法和应用
技术领域
1.本发明属于基因工程领域,具体地涉及一种毕赤酵母中iptg诱导的trna元件及其构建方法和应用。
背景技术:
2.细菌相关功能的结构基因常连在一起,形成一个基因簇。它们编码同一个代谢途径中不同的酶。大肠杆菌中乳糖分解代谢相关的三个基因,lacz、lacy、laca就是很典型的上述基因簇,他们分别编码β
‑
半乳糖苷酶(β
‑
galactosidase)、通透酶(permease)和乙酰基转移酶(transacetylase),催化乳糖的分解,产生葡萄糖和半乳糖。此外还有调控基因:操纵序列o(operator)、启动序列p(promoter),调节基因laci编码的lac阻遏物(lac repressor)。这一个完整的调节系统形成了一个共同的调节单位,这种调节单位就称为操纵子(operon)。操纵子的活性是由调节基因控制的。lac阻遏物是一种具有4个相同亚基的四级结构蛋白,每个亚基都有一个与诱导剂结合的位点。在没有乳糖存在时,lac操纵子处于阻遏状态,lac阻遏物,即laci蛋白能与操纵基因o结合,阻碍rna聚合酶与p序列结合,从而抑制转录启动。而当有诱导剂存在时,诱导剂可与阻遏蛋白结合,使阻遏蛋白构象发生变化,导致阻遏物从操纵基因o上解离下来,rna聚合酶不再受阻碍,启动子p启动反应开始发生转录。在这个操纵子体系中,真正的诱导剂并非乳糖本身。乳糖进入细胞,经β
‑
半乳糖苷酶催化,转变为半乳糖,即生理上的诱导剂。而在实验中,通常选用异丙基硫代半乳糖苷(iptg)作为诱导剂,iptg是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
3.转运rna(transfer rna),又称传送核糖核酸、转移核糖核酸,通常简称为trna,是一种由76
‑
90个核苷酸所组成的rna,其3'端可以在氨酰
‑
trna合成酶催化之下,接附特定种类的氨基酸。trna对于所有生物体的蛋白质合成都是必不可少的。蛋白质翻译的过程中,trna可借由自身的反密码子识别mrna上的密码子,将该密码子对应的氨基酸转运至核糖体合成中的多肽链上。在肽链生成过程中,第一个进入核糖体与mrna起始密码子结合的trna称为起始trna,其余trna参与肽链延伸,称为延伸trna,按照mrna上密码子的排列,携带特定氨基酸的trna依次进入核糖体。形成肽链后,trna即从核糖体释放出来,整个过程称为trna循环。被氨基酰化(或称带电)的trnas的比例是一个重要的生物学参数。trna的数量和带电trna的比例都会影响翻译的速度和效率。每个trna分子理论上只能与一种氨基酸接附,但是由于遗传密码的简并性,使得有多于一个以上的trna可以跟一种氨基酸接附。trna最早由robert m.bock成功结晶,之后陆续有人提出trna苜蓿叶状的二级结构。细菌细胞的总rna主要由rrna和trna组成,其中trna的含量高达20%。有研究表明,trna在细菌中的分布受生长和环境条件的调节,trna的产生与核糖体生物基因有关。然而,这种调控的分子机制仍不清楚。细菌中成熟的trna的分布被认为是由四个过程控制:trna基因的转录,trna前体的加工,trna前体的降解以及成熟trna的降解。
4.在真核生物中,rna聚合酶分为三种,其中iii型聚合酶负责转录所有已知的真核
trna基因。所有trna基因的转录是由一个内启动子(也称为ii型启动子)来调控的,此启动子位于trna基因的内部,可以由基因上游序列基序(最常见的是包括一个tata元件)进行调控。trna可以根据其反密码子来分类,在此分类基础上,酿酒酵母中的trna可分为42种。实际上,如果考虑trna本身序列上核苷酸的差异,trna的种类数目会更多。每种trna由分布在基因组上不同位置的几个拷贝的基因编码,一般认为细胞内trna的表达量与其基因拷贝数成正比,很少存在依赖转录因子的特定trna特异性调控机制。
5.分子生物学的中心法则描述了蛋白质合成的一般规律。在蛋白质翻译过程中,氨基酸的排列顺序由mrna上的三联密码子决定。在绝大多数生物体的核基因组中,共存在64个遗传密码子。除去三个终止密码子(taa,tag和tga),剩下的61个密码子编码20种标准氨基酸。除甲硫氨酸(met)和色氨酸(trp)只由一种密码子编码外,其它氨基酸都由二到六个密码子所编码,这些编码同一氨基酸的不同密码子称为同义密码子。同义密码子中,使用频率高的密码子被称为常见密码子或优化密码子,使用频率较低的密码子被称为稀有密码子或非优化密码子。一般来说,高表达基因更倾向于使用常见密码子,相应的,对应的trna丰度也更高。
6.毕赤酵母(pichia pastoris gs115)的基因操作简便,外源基因通过商业化质粒载体构建表达盒,可以通过同源重组的方式在基因组上整合并稳定遗传,不易丢失。对毕赤酵母表达系统的优化方式通常有增强启动子强度、提高重组蛋白分泌效率等。此外,密码子优化也是使用毕赤酵母系统表达重组蛋白过程中的一个常用步骤。极端的密码子优化固然可以提高翻译速度,却也会导致基因整体gc含量过低,在表达上产生各种各样的问题。通常来说,富含gc的密码子的mrna比具有较低gc含量的mrna更耐降解,因为它们具有更高的热力学稳定性,具有较长半衰期的mrna通常会产生更多蛋白质。但是毕赤酵母基因组中常见密码子第三位偏好at,这就导致了密码子优化在毕赤酵母中对mrna稳定性会朝着不同的方向改变。因此,在转录和翻译层面上不同时具备优势,是毕赤酵母系统常用密码子存在的问题。目前,不存在可诱导表达的毕赤酵母的trna元件。
技术实现要素:
7.本发明的目的是构建一项可诱导调控毕赤酵母的trna表达量的trna元件。本发明通过研究构建了iptg诱导的trna
‑
ile元件,可以解决毕赤酵母的trna表达的问题。因此本发明的第一个目的是提供一种毕赤酵母中iptg诱导的trna
‑
ile元件。本发明的第二个目的是提供所述的毕赤酵母中iptg诱导的trna
‑
ile元件的构建方法。本发明的第三个目的是提供所述的毕赤酵母中iptg诱导的trna
‑
ile元件的应用。
8.为了实现上述目的,本发明采用如下技术方案:
9.作为本发明的第一个方面,一种毕赤酵母中iptg诱导的trna元件,其是将乳糖操纵子的操纵序列laco1插入trna基因5’端,laco2插入trna基因3’端;两拷贝化后,连入载体pgapza的gap启动子序列之前,同时在载体gap启动子后插入n端带有nsv40序列的laci基因构建获得。
10.根据本发明,所述阻遏蛋白laci的序列如seq id no:26所示,所述laco2的序列如seq id no:30所示,所述laco1的序列如seq id no:27所示;nsv40序列如seq id no:29所示。
11.根据本发明,所述trna元件为trna
‑
ile元件。
12.作为本发明的第二个方面,一种毕赤酵母中iptg诱导的trna元件的构建方法,包括如下步骤:
13.步骤一、将序列如seq id no:29所示的nsv40序列插入序列如seq id no:26所示的阻遏蛋白laci基因的n端,插入载体pgapza的gap启动子序列下游,获得质粒pgapza
‑
sv40
‑
laci;
14.步骤二、将序列如seq id no:27所示的乳糖操纵子的操纵序列laco1插入trna基因5’端,获得基因p,以基因p为模板,扩增插入序列如seq id no:30所示的乳糖操纵子的操纵序列laco2,获得基因m;两拷贝化,获得基因n;然后将基因n插入质粒pgapza
‑
sv40
‑
laci的gap启动子序列之前,构建获得毕赤酵母中iptg诱导的trna元件。
15.根据本发明,步骤二是将序列如seq id no:27所示的乳糖操纵子的操纵序列laco1插入trnaile基因5’端,获得序列如seq id no:28所示的基因laco1
‑
trnaile,然后以基因laco1
‑
trnaile为模板,用序列如seq id no:23和seq id no:24所示的引物进行扩增,获得基因laco1o2
‑
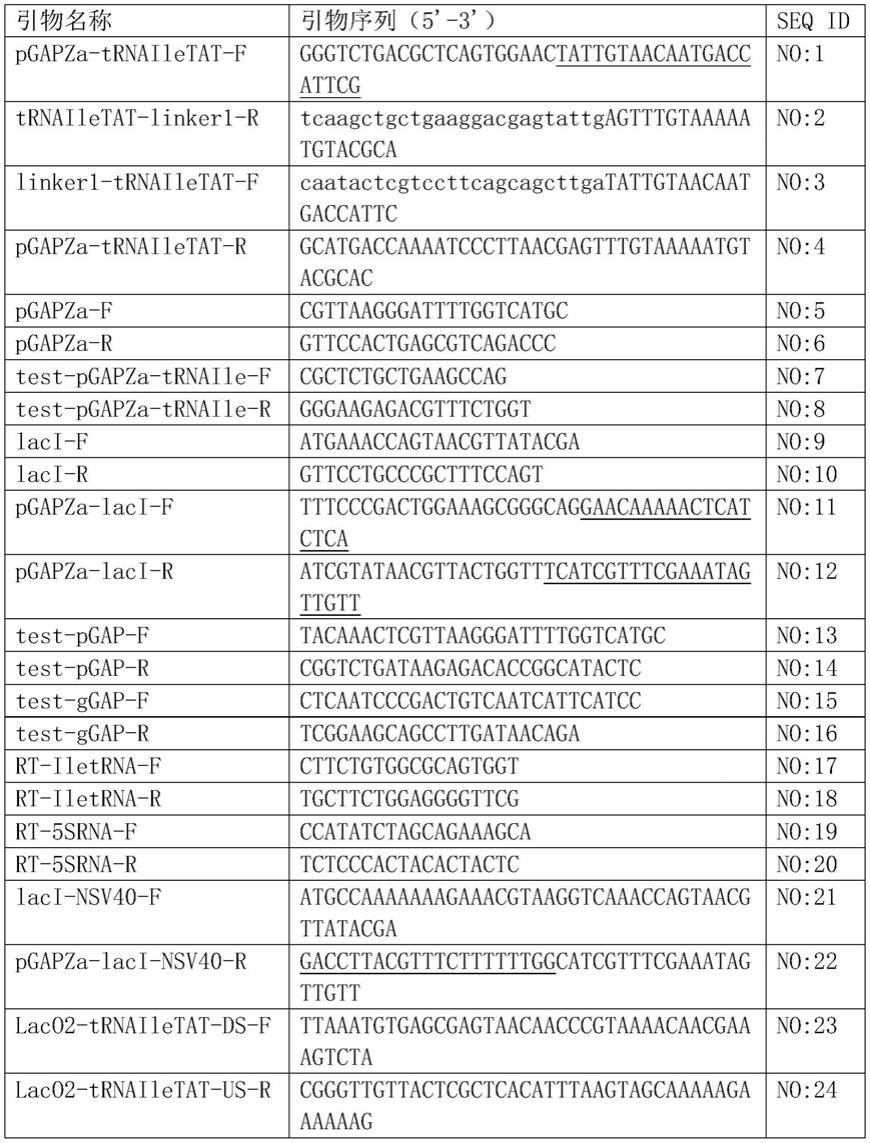
trnaile;接着,以基因laco1o2
‑
trnaile为模板,分别用如seq id no:1和seq id no:2以及如seq id no:3和seq id no:4所示的引物进行扩增,分别获得x片段与y片段;然后将pgapza
‑
sv40
‑
laci在gap启动子序列前线性化,并将x片段、y片段与线性化的质粒pgapza
‑
sv40
‑
laci进行连接转化,获得质粒pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci。
16.应当说明,以携带ile反密码子为tat的trna基因为例,所述毕赤酵母中iptg诱导的trna
‑
ile元件的构建方法包括:
17.步骤一、将序列如seq id no:29所示的nsv40序列插入序列如seq id no:26所示的阻遏蛋白laci基因的n端,获得基因nsv40
‑
laci,接着将载体pgapza在gap启动子序列之后线性化,与目的基因nsv40
‑
laci进行连接,转化进e.coli dh5a感受态后,获得质粒pgapza
‑
sv40
‑
laci;
18.步骤二、以序列如seq id no:27所示的laco1
‑
trnaile为模板,用序列如seq id no:23和seq id no:24所示的引物进行扩增,获得laco1o2
‑
trnaile;接着,以laco1o2
‑
trnaile为模板,分别用如seq id no:1和seq id no:2以及如seq id no:3和seq id no:4所示的引物进行扩增,分别获得x片段与y片段;然后将步骤一的质粒pgapza
‑
sv40
‑
laci在gap启动子序列前线性化,并将x片段、y片段与线性化的pgapza
‑
sv40
‑
laci进行连接转化,获得pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci,即毕赤酵母中iptg诱导的trna
‑
ile元件。
19.使用时,将质粒pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci线性化,并电转入毕赤酵母gs115,电转化复苏后,获得阳性基因组整合型多拷贝菌株gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci。
20.一种毕赤酵母中iptg诱导的trna
‑
ile元件,采用上述所述的构建方法构建获得。
21.一种毕赤酵母中iptg诱导的trna
‑
ile元件在毕赤酵母系统的密码子优化中的应用。
22.本发明的毕赤酵母中iptg诱导的trna元件,其有益效果:
23.能够通过小分子来调控一种trna的表达与否,然后通过大量表达这种trna能够逆转毕赤酵母之前的密码子偏好。
附图说明
24.图1为pgapza
‑
laci的质粒图谱。
25.图2为pgapza
‑
laco1trnaile
‑2‑
laci的质粒图谱。
26.图3为实施例4中构建的菌株,不同诱导条件时的trna的相对表达量的结果图。
27.图4为pgapza
‑
sv40
‑
laci的质粒图谱。
28.图5为pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci的质粒图谱。
29.图6为实施例8中构建的菌株,不同诱导条件时的trna的相对表达量的结果图。
30.图7为实施例12中northern blot的结果图。
31.图8为实施例13中trna跑测序胶的northern blot图。
具体实施方式
32.下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
33.1、本发明涉及的菌株和质粒来源
34.商品化质粒和菌株用于基因克隆。质粒pgapza,菌株e.coli dh5a,e.coli k12,pichia pastoris gs115均购自于invitrogen公司。
35.2、分子生物学试剂
36.所用到的2x taq pcr master mix、2x pfu pcr master、dna marker iii均购买于天根生化科技有限公司。质粒提取试剂盒购买于捷瑞生物工程有限公司。各种限制性核酸内切酶购买于thermo公司。无缝组装试剂盒one step cloning kit购自诺唯赞生物科技有限公司。dna荧光染料购自上海天能公司。
37.lysis buffer(裂解缓冲液):0.6m nacl,10mm edta,100mm tris ph 8.0,4%(w/v)sds,溶解于depc h2o中。
38.phe
‑
chl:苯酚:氯仿:异戊醇=25:24:1。
39.2x peg/nacl:10%peg
‑
8000,1m nacl,溶解于depc h2o中。
40.depc h2o:焦碳酸二乙酯处理过的水。它是一种rna酶抑制剂。
41.10x tbe buffer:108g tris,55g硼酸,7.44g na2edta,溶解于1l h2o,调节ph为8.3。
42.2x rna loading buffer(2x rna上样缓冲液):配制15ml时,需加入10ml去离子甲酰胺,750μl无菌甘油,3.5ml 40%甲醛,1.5ml 10x mops,3.75μl饱和溴酚蓝。需要注意的是,此buffer需要现配现用,如果用量少,可按比例缩小各试剂的用量。饱和溴酚蓝的配制方法为,40mg溴酚蓝粉末加入10ml水中。
43.temed(四甲基乙二胺),封闭液,洗涤液,streptavidin
‑
hrp(辣根过氧化物酶标记链霉亲和素),检测平衡液,显色液均购买于碧云天生物技术公司。
44.ssc,柠檬酸钠缓冲液;
45.sds,十二烷基磺酸钠。
46.μltrahyb buffer(杂交缓冲液)购买于thermo fisher scientific。
47.实施例12的带有生物素标记的rna探针合成于上海捷瑞生物工程有限公司,序列
为:
48.biotin
‑
ttcgaacccacgaccatcgcgttataagcacgatgcgctaaaccactg,seq id no:25。
49.3、培养基及培养条件
50.llb培养基:1%(w/v,下同)蛋白胨(tryptone),0.5%酵母粉(yeast extract),0.5%氯化钠(nacl),加入去离子水溶解。制备固体平板时再加入2%琼脂粉(agar)。121℃高压灭菌20min。
51.ypd培养基:2%(w/v,下同)蛋白胨(tryptone),1%酵母粉(yeast extract),2%葡萄糖(glucose),加入去离子水溶解。制备固体平板时再加入2%琼脂粉(agar)。配制时,葡萄糖单独配制成50%溶液,用0.22μm无菌滤膜过滤除菌,保存于4℃冰箱待用。蛋白胨和酵母粉按比例配制成溶液,121℃高压灭菌20min。使用时,葡萄糖按2%比例加入培养基中。
52.抗生素使用浓度:大肠杆菌:博莱霉素(zeocin)50μg/ml(需在llb培养基中使用)。毕赤酵母:博莱霉素(zeocin)100μg/ml。
53.培养条件:大肠杆菌于37℃条件下培养,毕赤酵母于30℃条件下培养。固体平板培养时需要倒置,液体培养时摇床转速为200rpm。
54.4、本发明所用的引物,如表1所示。
55.表1引物序列
[0056][0057]
实施例1
[0058]
以ncbi数据库pichia pastoris gs115(毕赤酵母gs115)的基因组序列为依据,依靠在线工具trnascan(http://lowelab.ucsc.edu/trnascan
‑
se/)确定所有的trna基因序列及拷贝数。以下挑选携带ile(异亮氨酸)反密码子为tat的trna基因为例,即trna
‑
ile的表达进行下一步研究。
[0059]
实施例2质粒pgapza
‑
laci的构建
[0060]
将阻遏蛋白laci导入载体pgapza。阻遏蛋白laci基因(1083bp)由e.coli k12菌株基因组作模板扩增而来,序列如seq id no:26所示:
[0061]
atgaaaccagtaacgttatacgatgtcgcagagtatgccggtgtctcttatcagaccgtttcccgcgtggtgaaccaggccagccacgtttctgcgaaaacgcgggaaaaagtggaagcggcgatggcggagctgaattacattcccaaccgcgtggcacaacaactggcgggcaaacagtcgttgctgattggcgttgccacctccagtctggccctgcacgcgccgtcgcaaattgtcgcggcgattaaatctcgcgccgatcaactgggtgccagcgtggtggtgtcgatggtagaacgaagcggcgtcgaagcctgtaaagcggcggtgcacaatcttctcgcgcaacgcgtcagtgggctgatcattaactatccgctggatgaccaggatgccattgctgtggaagctgcctgcactaatgttccggcgttatttcttgatgtctctgaccagacacccatcaacagtattattttctcccatgaagacggtacgcgactgggcgtggagcatctggtcgcattgggtcaccagcaaatcgcgctgttagcgggcccattaagttctgtctcggcgcgtctgcgtctggctggctggcataaatatctcactcgcaatcaaattcagccgatagcggaacgggaaggcgactggagtgccatgtccggttttcaacaaaccatgcaaatgctgaatgagggcatcgttcccactgcgatgctggttgccaacgatcagatggcgctgggcgcaatgcgcgccattaccgagtccgggctgcgcgttggtgcggatatctcggtagtgggatacgacgataccgaagacagctcatgttatatcccgccgttaaccaccatcaaacaggattttcgcctgctggggcaaaccagcgtggaccgcttgctgcaactctctcagggccaggcggtgaagggcaatcagctgttgcccgtctcactggtgaaaagaaaaaccaccctggcgcccaatacgcaaaccgcctctccccgcgcgttggccgattcattaatgcagctggcacgacaggtttcccgactggaaagcgggcag,
[0062]
seq id no:26。
[0063]
使用pgapza
‑
laci
‑
f/pgapza
‑
laci
‑
r引物pcr线性化,pcr产物通过切胶回收。将目的基因laci与线性化载体pgapza无缝组装,化学转化进e.coli dh5a感受态复苏后均匀涂布于含zeocin抗生素的llb平板上,过夜培养长出单菌落后,挑取单菌落进行菌落pcr验证。质粒构建成功后命名为pgapza
‑
laci,质粒图谱如图1所示。
[0064]
实施例3质粒pgapza
‑
laco1trnaile
‑2‑
laci的构建
[0065]
将乳糖操纵子的操纵序列laco1插入trnaile5’端,由捷瑞公司进行合成,获得laco1
‑
trnaile。其中,乳糖操纵子的操纵序列laco1的序列如seq id no:27所示:
[0066]
tgtgtggaattgtgagcggataacaatttcacaca,seq id no:27。
[0067]
laco1
‑
trnaile的基因序列如seq id no:28所示:
[0068]
tattgtaacaatgaccattcgtttcatagttgcagaacattataaaattcctcaactatgctcgttatatgctagtcaataaaaaacatggcatgagtgatgtgtggaattgtgagcggataacaatttcacacagcttctgtggcgcagtggtttagcgcatcgtgcttataaacgtttaacacgtctatgttcaaagcacagtcgtttagaaacgcgatggtcgtgggttcgaacccctccagaagcattgcttacttttttctttttgctactaaaacaacgaaagtctaggcctattgtctcctgtttcacttaacatgtttaaagtgcgtacatttttacaaact,seq id no:28。
[0069]
接着,用overlap pcr的方法将laco1
‑
trnaile扩增为两拷贝,所用引物为pgapza
‑
trnailetat
‑
f/trnailetat
‑
linker1
‑
r,linker1
‑
trnailetat
‑
f/pgapza
‑
trnailetat
‑
r,获得基因laco1
‑
trnaile
‑
2。
[0070]
用pcr的方法线性化载体pgapza
‑
laci,所用引物为pgapza
‑
f/pgapza
‑
r。pcr产物通过切胶回收,将laco1
‑
trnaile
‑
2与pgapza
‑
laci载体用无缝组装试剂盒进行连接,化学转化进e.coli dh5a感受态,复苏后均匀涂布于含zeocin抗生素的llb平板上,过夜培养长出单菌落后,挑取单菌落进行菌落pcr验证。验证引物为test
‑
pgapza
‑
trnaile
‑
f/test
‑
pgapza
‑
trnaile
‑
r,将验证后的阳性转化子液体培养过夜后,用试剂盒提取质粒送由金唯智公司测序,质粒构建成功后命名为pgapza
‑
laco1trnaile
‑2‑
laci,质粒图谱如图2所示。
[0071]
实施例4酵母菌株gs115
‑
pgapza
‑
laco1trnaile
‑2‑
laci的构建
[0072]
质粒pgapza
‑
laco1trnaile
‑2‑
laci用avrii在pgap启动子内部酶切线性化,回收后电转入毕赤酵母gs115。为了得到高拷贝菌株,电转化复苏后,将其涂布于含高浓度zeocin抗生素的ypd平板上(其中,抗生素zeocin终浓度分别为0.5mg/ml,0.7mg/ml,1mg/ml)。倒置培养两天,长出肉眼可见的菌落后,挑取单菌落于ep管中培养两天。然后提取基因组进行pcr验证。验证引物为test
‑
pgap
‑
f/test
‑
ggap
‑
r,test
‑
ggap
‑
f/test
‑
pgap
‑
r。筛选基因组整合型多拷贝菌株gs115
‑
pgapza
‑
laco1trnaile
‑2‑
laci。保存于20%甘油溶液中,冻存于
‑
80℃。
[0073]
实施例5 gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
nsv40
‑
laci的培养及诱导表达
[0074]
挑取转化子15#号菌株于5ml ypd液体培养基中,30℃摇床培养两天后,转接到含zeocin抗生素的50ml ypd液体培养基中,待菌体长到对数中期时,加入不同浓度的iptg(2mm、5mm)进行诱导,10h后离心收菌进行rna的提取。
[0075]
实施例6毕赤酵母总rna与小rna的提取
[0076]
1、总rna的提取
[0077]
1)将实施例5培养好的菌液转移至离心管中,5000rpm离心10min,弃去上清,收集沉淀。
[0078]
2)将沉淀转移至研钵中,加入适量液氮研磨,直至菌体被研磨成细小均一的粉末状态。
[0079]
3)将粉末收集到ep管中,加入450μl lysis buffer(裂解缓冲液)与450μl phe
‑
chl(苯酚
‑
氯仿),充分漩涡混匀。
[0080]
4)12000rpm,4℃离心10min。
[0081]
5)将上层溶液转移至含有450μl phe
‑
chl的新ep管中,漩涡混匀。
[0082]
6)12000rpm,4℃离心10min。
[0083]
7)小心吸取上层溶液至新的ep管中,尽量不要吸到下层溶液,加入上层溶液的1/10体积的3m naoac和2.5倍体积的冰冷的95%乙醇,混合均匀。放置于
‑
80℃过夜(至少
‑
20℃,1h)以增加产量。
[0084]
8)12000rpm,4℃离心10min。弃去上清。
[0085]
9)用80%的乙醇洗涤沉淀一次,放置一段时间挥干乙醇。
[0086]
10)用适量的rnase
‑
free water溶解沉淀。nano
‑
300测定浓度。
[0087]
2、小rna的提取
[0088]
1)取大约250μg的总rna溶液与等体积的2x peg/nacl混合,冰浴30min。
[0089]
2)12000rpm,4℃离心10min。
[0090]
3)将上层溶液转移至新的ep管中,加入1/10体积的3m naoac和3倍体积的100%冰冷的乙醇,混合均匀。放置于
‑
80℃过夜(至少
‑
20℃,1h)以增加产量。
[0091]
4)12000rpm,4℃离心10min。弃去上清。
[0092]
5)用80%的乙醇洗涤沉淀一次,放置一段时间挥干乙醇。
[0093]
6)用适量的rnase
‑
free water(无rna酶的水)溶解沉淀。nano
‑
300测定浓度。
[0094]
注:上述步骤所用ep管、吸头等都应是无rna酶的,所用其他容器用depc h20擦拭处理。
[0095]
以5s rna为内参基因进行rt
‑
qpcr测定所导入的trna
‑
ile在不同诱导条件时的相对表达量。所用引物为rt
‑
iletrna
‑
f/rt
‑
iletrna
‑
r,rt
‑
5srna
‑
f/rt
‑
5srna
‑
r。
[0096]
结果如图3所示。其中,wt组为gs115野生型菌株对照组,2mm iptg、5mm iptg分别为添加有2mm、5mm的iptg的菌株,15#为不加iptg的菌株。
[0097]
结果显示,不加iptg的菌株trna水平比wt的trna表达高许多,表明使用pgapza
‑
laco1trnaile
‑2‑
laci系统没有达到有效地阻遏trna表达。
[0098]
实施例7质粒pgapza
‑
sv40
‑
laci的构建
[0099]
在laci基因n端添加核定位序列nsv40。nsv40序列为ccaaaaaagaaacgtaaggtc(seq id no:29),因其比较短,所以在扩增laci基因时,直接将其序列设计在引物上,这样通过一步克隆我们就能将nsv40基因插入在laci基因的n端。
[0100]
laci基因用laci
‑
nsv40
‑
f/laci
‑
r引物进行扩增,获得目的基因nsv40
‑
laci。
[0101]
质粒pgapza用pgapza
‑
laci
‑
f/pgapza
‑
laci
‑
nsv40
‑
r引物pcr线性化。pcr产物通过切胶回收,将目的基因nsv40
‑
laci与线性化载体pgapza无缝组装,化学转化进e.coli dh5a感受态,复苏后均匀涂布于含zeocin抗生素的llb平板上,过夜培养长出单菌落后,挑取单菌落进行菌落pcr验证。质粒构建成功后命名为pgapza
‑
sv40
‑
laci,质粒图谱如图4所示。
[0102]
实施例7质粒pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci的构建
[0103]
为了加强laci的阻遏效果,在trna转录终止位点之后又插入了第二段laci结合序列laco2。其中,laco2序列为ttaaatgtgagcgagtaacaacccg(seq id no:30)。
[0104]
使用引物laco2
‑
trnailetat
‑
ds
‑
f/laco2
‑
trnailetat
‑
us
‑
r,以laco1
‑
trnaile(其序列如seq id no:28所示)为模板进行pcr,得到laco1o2
‑
trnaile序列。然后使用引物pgapza
‑
trnailetat
‑
f/trnailetat
‑
linker1
‑
r,linker1
‑
trnailetat
‑
f/pgapza
‑
trnailetat
‑
r,以laco1o2
‑
trnaile为模板分别会扩增出420bp与400bp的片段,将其分别命名为x片段与y片段。
[0105]
用pgapza
‑
f/pgapza
‑
r将载体pgapza
‑
sv40
‑
laci线性化。通过多片段一步克隆试剂盒将线性化pgapza
‑
sv40
‑
laci、x片段、y片段进行连接转化。测序无误后将质粒命名为pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci。质粒图谱如图5所示。
[0106]
实施例8酵母菌株gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci的构建
[0107]
质粒pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci用avrii在pgap启动子内部酶切线性化,回收后电转入毕赤酵母gs115。为了得到高拷贝菌株,电转化复苏后,将其涂布于含高浓度zeocin抗生素的ypd平板上(其中,抗生素zeocin终浓度分别为0.5mg/ml,0.7mg/ml,1mg/ml)。倒置培养两天,长出肉眼可见的菌落后,挑取单菌落于ep管中培养两天。然后提取基因组进行pcr验证。验证引物为test
‑
pgap
‑
f/test
‑
ggap
‑
r,test
‑
ggap
‑
f/test
‑
pgap
‑
r。筛选基因组整合型多拷贝菌株gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci。保存于20%甘油溶液中,冻存于
‑
80℃。
[0108]
实施例9 gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci的培养及诱导表达
[0109]
挑取转化子4#号菌株于5ml ypd液体培养基中,30℃摇床培养两天后,转接到含zeocin抗生素的50ml ypd液体培养基中,待菌体长到对数中期时,加入不同浓度的iptg(0.5mm、1mm、2mm、5mm)进行诱导,10h后离心收菌进行rna的提取。
[0110]
实施例10毕赤酵母总rna与小rna的提取
[0111]
1.总rna的提取
[0112]
(1)将实施例9培养好的菌液转移至离心管中,5000rpm离心10min,弃去上清,收集沉淀。
[0113]
(2)将沉淀转移至研钵中,加入适量液氮研磨,直至菌体被研磨成细小均一的粉末状态。
[0114]
(3)将粉末收集到ep管中,加入450μl lysis buffer(裂解缓冲液)与450μl phe
‑
chl(苯酚
‑
氯仿),充分漩涡混匀。
[0115]
(4)12000rpm,4℃离心10min。
[0116]
(5)将上层溶液转移至含有450μl phe
‑
chl的新ep管中,漩涡混匀。
[0117]
(6)12000rpm,4℃离心10min。
[0118]
(7)小心吸取上层溶液至新的ep管中,尽量不要吸到下层溶液,加入上层溶液的1/10体积的3m naoac和2.5倍体积的冰冷的95%乙醇,混合均匀。放置于
‑
80℃过夜(至少
‑
20℃,1h)以增加产量。
[0119]
(8)12000rpm,4℃离心10min。弃去上清。
[0120]
(9)用80%的乙醇洗涤沉淀一次,放置一段时间挥干乙醇。
[0121]
(10)用适量的rnase
‑
free water溶解沉淀。nano
‑
300测定浓度。
[0122]
2.小rna的提取
[0123]
(1)取大约250μg的总rna溶液与等体积的2x peg/nacl混合,冰浴30min。
[0124]
(2)12000rpm,4℃离心10min。
[0125]
(3)将上层溶液转移至新的ep管中,加入1/10体积的3m naoac和3倍体积的100%冰冷的乙醇,混合均匀。放置于
‑
80℃过夜(至少
‑
20℃,1h)以增加产量。
[0126]
(4)12000rpm,4℃离心10min。弃去上清。
[0127]
(5)用80%的乙醇洗涤沉淀一次,放置一段时间挥干乙醇。
[0128]
(6)用适量的rnase
‑
free water(无rna酶的水)溶解沉淀。nano
‑
300测定浓度。
[0129]
注:上述步骤所用ep管、吸头等都应是无rna酶的,所用其他容器用depc h20擦拭处理。
[0130]
实施例11 rt
‑
qpcr比较不同iptg诱导浓度下trna
‑
ile的表达量
[0131]
以5s rna为内参基因进行rt
‑
qpcr测定所导入的trna
‑
ile在不同诱导条件时的相对表达量。所用引物为rt
‑
iletrna
‑
f/rt
‑
iletrna
‑
r,rt
‑
5srna
‑
f/rt
‑
5srna
‑
r。结果如图6所示。其中,wt组为gs115野生型菌株对照组,0.5mm iptg、1mm iptg、2mm iptg、5mm iptg分别为添加有0.5mm、1mm、2mm、5mm的iptg的菌株,4#为不加iptg的菌株。
[0132]
结果显示,不加iptg的菌株trna水平与wt相当,表明使用laco1o2双阻遏序列系统可以成功阻遏trna表达。用不同浓度的iptg诱导后,1mm或以上iptg可以显著诱导trna
‑
ile表达。
[0133]
实施例12 northern blot实验
[0134]
1.配胶:一般1.5mm厚度的凝胶需配制10ml液体胶;1.0mm厚度的凝胶需配制5ml液体胶。根据所分离的rna分子的大小配制不同浓度的凝胶。本实验配制的为6%的胶。
[0135]
(1)用depc h2o仔细擦洗制胶所用的玻璃板,胶架以及梳子,再用水冲洗一遍,将
其安装至胶架上,加入去离子水检漏。。
[0136]
(2)等待2min胶板不漏后,倒出胶板中的水,用吸水纸吸干残留的水。
[0137]
(3)配制20ml的凝胶时,称取10g尿素于小烧杯中,加入3ml 40%acr
‑
bis(19:1),2ml 10x tbe buffer,补depc h2o至20ml,加热搅拌至尿素颗粒完全溶解。
[0138]
(4)加入200μl 10%aps(过硫酸铵溶液),在通风橱中加入8μl temed。搅拌均匀,在液态胶凝固之前将其小心注入玻璃板中,按需求插入10或15孔的梳子。
[0139]
(5)室温下放置一段时间待其凝固。
[0140]
2.rna样品制备:取适量rna样品加入等体积的2x rna loading buffer(2x rna上样缓冲液),置于95℃金属浴加热5min,立即置于冰上冷却5min。
[0141]
3.电泳:
[0142]
(1)将电泳胶固定在电泳槽中,加入1l 1x tbe buffer,使液面完全没过电泳胶,拔掉梳子,按需求点样。
[0143]
(2)盖上电泳槽的盖子,注意电极正负。调节电压为300v,开始电泳,待最下面的蓝色条带完全跑出后结束电泳。小心拆下凝胶,进行下一步实验。
[0144]
4.半干转膜
[0145]
(1)将电泳胶在预冷的0.5x tbe buffer中浸泡15min。
[0146]
(2)根据胶的大小裁剪滤纸和hybond
‑
n+膜(带正电尼龙膜)。膜的大小略大于胶。膜和滤纸都用0.5x tbe buffer润湿。
[0147]
(3)将上述物品按照从下到上滤纸
‑
膜
‑
胶
‑
滤纸的顺序放置在半干转膜仪上,盖上仪器的盖子,注意正负极。
[0148]
(4)恒流400ma转膜1h,注意电压不能超过25v,否则会损坏仪器。
[0149]
5.紫外交联
[0150]
将转膜完成的膜用镊子夹出,放入紫外交联仪中交联1min。
[0151]
6.探针杂交
[0152]
将膜放置于杂交管中,注意正面朝向管子的内部,加入适量mltrahyb buffer,杂交管置于旋转杂交炉中,42℃预杂交2h。之后按照千分之一的比例加入带有生物素标记的rna探针,42℃杂交过夜。
[0153]
7.洗膜:杂交完成的膜用洗膜缓冲液(0.5x ssc和0.2%sds)洗三次,每次20min,洗膜依然在42℃的旋转杂交炉中完成。
[0154]
8.化学发光检测
[0155]
(1)37
‑
50℃水浴溶解封闭液和洗涤液,确保这两种溶液中均无沉淀。
[0156]
(2)取一合适的容器加入15ml封闭液,再放入尼龙膜,在侧摆摇床或水平摇床上缓慢摇动15min。
[0157]
(3)取7.5μl streptavidin
‑
hrp加入到15ml封闭液中(1:2000稀释)。
[0158]
(4)弃去封闭液,加入步骤(3)配制溶液,在侧摆摇床或水平摇床上缓慢摇动15min。
[0159]
(5)加入1x洗涤液15
‑
20ml,漂洗1min。
[0160]
(6)弃去洗涤液,加入新的洗涤液15
‑
20ml,在侧摆摇床或水平摇床上洗涤5min。
[0161]
(7)重复步骤(6)三次。
[0162]
(8)将尼龙膜转移至新的容器中,加入20ml检测平衡液,在侧摆摇床或水平摇床上缓慢摇动5min。
[0163]
(9)将尼龙膜用滤纸吸干,放置在化学发光仪黑箱中,均匀滴上配制好的显色液,打开软件,拍摄图片。
[0164]
分别从保种管中接取gs115 wt以及gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
nsv40
‑
laci于5ml ypd液体培养基中,30℃摇床培养两天后,转接到含0.1mg/ml的zeocin抗生素的50ml ypd液体培养基中,待菌体长到对数中期时,加入不同浓度的iptg(0.5mm、1mm、2mm、5mm)进行诱导,10h后离心收菌进行rna的提取。分别取1μg的总rna按照上述步骤进行northern blot,结果如图7所示。
[0165]
结果说明:我们构建的菌株gs115
‑
pgapza
‑
laco1o2trnaile
‑2‑
sv40
‑
laci在iptg诱导条件下可以顺利表达trna
‑
ile。
[0166]
实施例13测序胶实验
[0167]
正常体内的trna会携带相应的氨基酸,在碱性条件下,氨基酸会脱去,因此分子量会减小。通过测序胶实验,经过碱性处理的trna样品会出现两个条带,即一个携带氨基酸的稍大条带,和一条脱去氨基酸的稍小的条带。因此,此实验可以确定我们所导入的trna是否能够正常携带氨基酸。
[0168]
1.取酸性条件下提取的wt总rna 1μg于rnase
‑
free ep管中,加入10μl 50mm tris
‑
hcl(ph=9.0),混匀后放入金属浴37℃孵育1h。
[0169]
2.分别取酸性条件下提取的wt,4#,4#
‑
2mm iptg总rna 1μg于rnase
‑
free ep管中,加depc h2o补至10μl,混匀。
[0170]
3.上述5个样品进行酸性测序胶northern blot,结果如图8所示。
[0171]
结果显示,wt总rna经过碱处理后,的确产生了一条稍小的条带,而我们构建的菌株,其条带与未经处理的wt保持一致,说明导入的trna可以正常携带氨基酸。
[0172]
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1