一种手性吲哚酮类衍生物及其合成方法
本发明属于医药化工技术领域。更具体地,涉及一种手性吲哚酮类衍生物及其合成方法。
背景技术:
2-吲哚酮结构广泛存在于天然产物、药物和生物活性分子中,具有抗肿瘤、抗炎、促生长、抗帕金森等药理作用。如l-oxindolylalanine为l-色氨酸的代谢产物,具有良好的抑菌活性;fr900452和maremycinsb是由海洋链霉菌streptomycessp.b9173中产生的生物碱类化合物,对淋巴癌细胞具有一定的抑制活性;a.scala等人(bioorgmedchem.,2014,22(3):1063-9)报道了一类对利什曼原虫原虫的具有抗增殖作用的3-取代-2-吲哚酮化合物;从鼠尾草根中分离得到的prioline,可用于治疗扁桃体炎、咽炎、肺结核和细菌性痢疾;生物碱化合物ammosamideb和piperumbellactama具有相似的氧化吲哚结构,具有抗肿瘤活性和α-葡萄糖苷酶抑制作用。可见,构建结构多样性的2-吲哚酮化合物具有潜在的应用价值。
现有报道合成手性吲哚酮的方法,主要集中在对前手性吲哚的不对称加成反应,如yuan等报道了在二苯基乙二胺手性催化剂下,吲哚酮与烯酮的不对称michael加成反应构建手性3-取代2-吲哚酮吲哚酮衍生物,但该反应只有在伯胺和d-boc保护的氨基酸共同作用下,才能有效地催化该反应的进行,且反应路线过长(organicletters,2015.17,11,2732–2735);paolomelchiorre等报道利用新型双功能伯胺硫脲催化剂实现了不饱和烯烃和吲哚酮的1,4michael加成反应,但该反应只有伯胺和硫脲催化剂协同活化同时作用,才能立体选择性的促进c-c键的形成,且该底物适用范围不广,反应路线过长(chemistryaeuropeanjournal,2010,15(32),7846-7849)。综上,现有的合成方法中,存在合成路线过长、底物适用范围有限等不足之处。因此,发展简洁、高效率的合成手性吲哚酮类衍生物的方法具有重要的意义。
技术实现要素:
本发明要解决的技术问题是克服现有手性吲哚酮类衍生物合成方法的缺陷和不足,提供一种手性吲哚酮类衍生物的合成方法。该合成方法简单、反应快、底物适用性广,为制备手性3-取代-2-吲哚酮衍生物提供了一种新的方法,具有较大应用前景。
本发明的另一目的是提供一种手性吲哚酮类衍生物。
本发明上述目的通过以下技术方案实现:
一种手性吲哚酮类衍生物的合成方法,以重氮酰胺、醛和烯胺为原料,加入吸水剂,以取代苯胺、钯(ii)二聚体和手性磷酸为催化剂,在有机溶剂中,反应得到式(i)所示手性吲哚酮类衍生物,
重氮酰胺的结构式为
烯胺的结构式为
其中,r1选自c6-c10芳基或c1-c6烷基;
r2选自氢、c1-c6烷基、c1-c6烷氧基或卤素;
r3选自氢、c1-c3烷基;
r4选自c6-c10芳基、c4-c8杂环芳基或c2-c8炔基;
ar选自c6-c10芳基或c4-c8杂环芳基;
其中,所述c2-c8炔基上的氢原子任选地被下组取代基取代:卤素、c1-c3烷氧基、c1-c3卤代烷基、氰基、硝基、酯基、c6-c10芳基或c4-c8杂环芳基;
所述c1-c6烷基上的氢原子任选地被下组取代基取代:卤素、c1-c3卤代烷基、氰基、硝基、酯基或c1-c6烷氧基;
所述c6-c10芳基上的氢原子任选地被下组取代基取代:卤素、c1-c3卤代烷基、氰基、硝基、酯基、c1-c6烷氧基或c1-c6烷基。
本发明通过一锅法反应高效构建手性吲哚酮类衍生物,该合成方法具有操作简单、反应快、底物适应性广等优势。本发明以重氮酰胺、醛和烯胺为原料,以取代苯胺、钯(ii)二聚体和手性磷酸组成催化体系,在有机溶剂中,醛在取代苯胺的作用下形成亚胺,然后与烯胺在手性磷酸作用下产生高亲电性的亚胺盐中间体,而重氮酰胺在钯(ii)二聚体作用下,形成金属卡宾,随后发生分子内的c-h活化反应形成五元环结构的两性离子对中间体,该中间体被亚胺盐中间体捕捉形成烯胺产物,再经过水解形成手性吲哚酮类衍生物。本发明反应机理如下:
其中,本发明以取代苯胺、钯(ii)二聚体和手性磷酸组成的催化体系中,所述手性磷酸为有机分子催化剂,其作用为促进亚胺盐中间体形成,并控制反应的立体选择性,进而更高效地合成手性吲哚酮类衍生物;所述钯(ii)二聚体为金属催化剂,其作用是分解重氮酰胺形成五元环结构的两性离子对中间体;所述取代苯胺为有机分子催化剂,其作用是活化醛。
优选地,所述钯(ii)二聚体为烯丙基氯化钯二聚体、2-甲基烯丙基氯化钯二聚体、氯化丁烯钯二聚体或氯化肉桂基钯二聚体。
更优选地,所述钯(ii)二聚体为烯丙基氯化钯二聚体。
优选地,所述取代苯胺为对氯苯胺、间氯苯胺、邻氯苯胺、对甲苯胺、邻甲苯胺、间甲苯胺、对甲氧基苯胺、间甲氧基苯胺、邻甲氧基苯胺、对溴苯胺、邻溴苯胺、间溴苯胺、对硝基苯胺、邻硝基苯胺或间硝基苯胺。
优选地,所述催化剂中,取代苯胺、钯(ii)二聚体和手性磷酸的摩尔比为1~10:0.5:1。
更优选地,所述催化剂中,取代苯胺、钯(ii)二聚体和手性磷酸的摩尔比为6:0.5:1。
优选地,所述原料中,醛、重氮酰胺和烯胺的摩尔比为1.0:1.2~1.8:1.2。
更优选地,所述原料中,醛、重氮酰胺和烯胺的摩尔比为1.0:1.5:1.2。
优选地,所述吸水剂为分子筛。常用的分子筛有
更优选地,所述吸水剂为
优选地,以醛的摩尔量为基准,所述吸水剂的投料量为50~200mg/mmol。
优选地,所述反应的温度为-30℃~0℃。
更优选地,所述反应的温度为0℃。
优选地,所述反应的时间为1~2h。
更优选地,所述反应的时间为1h。
优选地,所述有机溶剂为二氯甲烷、二氯乙烷、甲苯或甲基叔丁基醚中的一种或多种。
优选地,所述有机溶剂与醛的摩尔体积比为0.5~1.0ml:0.1mmol。
更优选地,所述有机溶剂与醛的摩尔体积比为1.0ml:0.1mmol。
在一个具体的实施方案中,本发明手性吲哚酮类衍生物的合成方法,包括以下步骤:以醛的摩尔量为基准,按摩尔比为重氮酰胺:醛:烯胺:取代苯胺:钯(ii)二聚体:手性磷酸=1.5:1.0:1.2:0.6:0.05:0.1,称取原料。将醛、取代苯胺、钯(ii)二聚体、手性磷酸、吸水剂溶于有机溶剂,配置混合溶液a;将重氮酰胺与烯胺溶于有机溶剂配置混合溶液b;在0℃下,将混合溶液b在3h内通过蠕动泵加入混合溶液a中;同时剧烈搅拌;混合溶液b加入完毕后,0℃下继续搅拌1h,直至重氮化合物消耗完全;将粗产物柱层析得到纯产品式(i)所示手性吲哚酮类衍生物。
本发明还保护一种手性吲哚酮类衍生物,具有式(i)所示结构:
其中,r1选自c6-c10芳基或c1-c6烷基;
r2选自氢、c1-c6烷基、c1-c6烷氧基或卤素;
r3选自氢或c1-c3烷基;
r4选自c6-c10芳基、c4-c8杂环芳基或c2-c8炔基;
ar选自c6-c10芳基或c4-c8杂环芳基;
其中,所述c2-c8炔基上的氢原子任选地被下组取代基取代:卤素、c1-c6烷氧基、c1-c3卤代烷基、氰基、硝基、酯基、c6-c10芳基或c4-c8杂环芳基;
所述c1-c6烷基上的氢原子任选地被下组取代基取代:卤素、c1-c3卤代烷基、氰基、硝基、酯基或c1-c3烷氧基;
所述c6-c10芳基上的氢原子任选地被下组取代基取代:卤素、c1-c3卤代烷基、氰基、硝基、酯基、c1-c3烷氧基或c1-c6烷基。
其中,所述c4-c8杂环芳基为含氧、硫或氮原子的c4-c8杂环芳基。
优选地,r1为甲基、乙基或苄基。
优选地,r2为氢、甲基、乙基、丙基、甲氧基、乙氧基、氟、氯或溴。
更优选地,r2为氢、5-甲基、5-甲氧基、5-氟、5-氯、5-溴、4-甲基或6-甲基。
优选地,r3为氢、甲基或乙基。
优选地,r4为苯乙炔基、2-甲基苯乙炔基、4-甲基苯乙炔基、4-甲氧基苯乙炔基、2-氟苯乙炔基,3-氯苯乙炔基、4-氯苯乙炔基、4-三氟甲基苯乙炔基、4-甲氧甲酰基苯乙炔基、4-氰基苯乙炔基、2-噻吩乙炔基、3-噻吩乙炔基、庚炔基、2-甲基苯基、3-甲基苯基、4-甲基苯基、4-甲氧基苯基、4-溴苯基、4-碘苯基、4-硝基苯基、2-噻吩基、2-萘基、3-吲哚基、2-呋喃基、3-苯并噻吩基或2-(n-甲基吡咯)基。
更优选地,r4为苯乙炔基、4-甲基苯乙炔基、4-甲氧基苯乙炔基、4-氯苯乙炔基或4-甲氧甲酰基苯乙炔基。
优选地,ar选自苯基、卤代苯基、c1-c6烷基苯基或杂环芳基。
更优选地,ar为苯基、4-氟苯基、4-氯苯基、4-甲基苯基或2-呋喃基。
本发明还提出了一种根据上述合成方法进一步衍生所得到的光学活性吲哚酮衍生物,所述光学活性吲哚酮衍生物的结构如式(ii)和式(iii)所示,
本发明合成方法可简单高效地制备得到手性吲哚酮类衍生物,还可以通过进一步的化学转化构建式(ii)和式(iii)所示光学活性吲哚酮衍生物。此外,该化合物的稠合吲哚酮结构单元具有抗肿瘤、抗炎、促生长等药理作用,具有较高的应用价值。
与现有技术相比,本发明具有以下有益效果:
本发明提供一种手性吲哚酮类衍生物的合成方法及其合成的手性吲哚酮类衍生物。本发明所述合成方法以重氮酰胺、醛和烯胺为原料,加入吸水剂,以取代苯胺、钯(ii)二聚体和手性磷酸组成催化体系,在有机溶剂中,一步高效构建手性吲哚酮类衍生物,该合成方法操作简单、反应快、底物适应性广,且具有产率高、非对映选择性和对映选择性高等优点。本发明合成方法可简单高效地合成具有两个季碳中心的手性吲哚酮类衍生物,还可以通过进一步的化学转化构建具有光学活性吲哚酮衍生物。因此,本发明在医药化工领域具有良好的应用前景。
附图说明
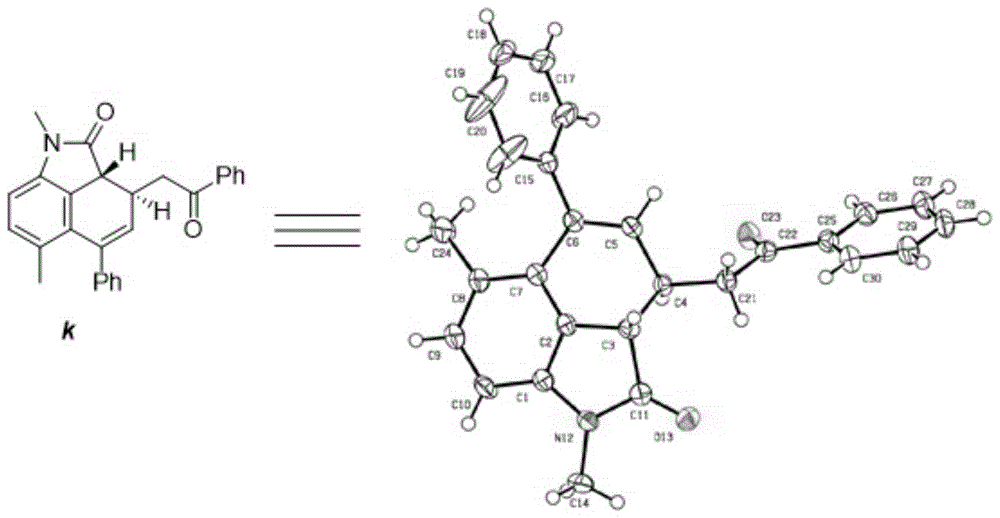
图1是本发明产物k的单晶衍射图;
图2是本发明产物l的单晶衍射图;
图3为实施例1所得产物的1hnmr图;
图4为实施例1所得产物的13cnmr图;
图5为实施例2所得产物的1hnmr图;
图6为实施例2所得产物的13cnmr图;
图7为实施例3所得产物的1hnmr图;
图8为实施例3所得产物的13cnmr图;
图9为实施例4所得产物的1hnmr图;
图10为实施例4所得产物的13cnmr图;
图11为实施例4所得产物的19fnmr图;
图12为实施例5所得产物的1hnmr图;
图13为实施例5所得产物的13cnmr图;
图14为实施例6所得产物的1hnmr图;
图15为实施例6所得产物的13cnmr图;
图16为实施例7所得产物的1hnmr图;
图17为实施例7所得产物的13cnmr图;
图18为实施例8所得产物的1hnmr图;
图19为实施例8所得产物的13cnmr图;
图20为实施例9所得产物的1hnmr图;
图21为实施例9所得产物的13cnmr图;
图22为实施例10所得产物的1hnmr图;
图23为实施例10所得产物的13cnmr图;
图24为实施例11所得产物的1hnmr图;
图25为实施例11所得产物的13cnmr图;
图26为实施例12所得产物的1hnmr图;
图27为实施例12所得产物的13cnmr图。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
其中,本发明原料化合物重氮酰胺的合成方法为:以n-甲基重氮乙酰对甲苯胺为例,将n-甲基苯胺(0.4g,3.8mmol)和二异丙基乙基胺(0.66ml,3.8mmol)溶解于二氯甲烷(20.0ml)中,在冰水浴下,缓慢滴加含溴乙酰溴(0.34ml,3.8mmol)的二氯甲烷(5.0ml)溶液。滴加完后升至室温并搅拌2h。反应完毕后,加入1n盐酸(约20ml)淬灭反应,分液,无水硫酸钠干燥有机相,减压除去溶剂得粗产物。粗产物不经纯化,直接和双对甲苯磺酰肼(3.2g,9.5mmol)溶解于四氢呋喃(20.0ml)中,冰水浴下,滴加dbu(2.7ml,18.0mmol)。继续在冰水浴下搅拌10min后,加入20ml饱和碳酸氢钠水溶液淬灭反应,再加入20ml乙酸乙酯稀释后,分液,无水硫酸钠干燥,减压除去溶剂得粗产物。粗产物柱层析分离纯化(乙酸乙酯:石油醚=1:10~1:5)得0.4g橙黄色油状产物,即为n-甲基重氮乙酰对甲苯胺,两步反应收率为60%。
实施例1化合物a的合成
将苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(400mhz,cdcl3)δ8.02(d,j=7.4hz,2h),7.59(t,j=7.3hz,1h),7.49(t,j=7.6hz,2h),7.34(s,1h),7.21(dd,j=6.3,3.6hz,5h),7.12(d,j=7.8hz,1h),6.74(d,j=7.9hz,1h),3.92(dd,j=13.8,6.3hz,1h),3.72–3.68(m,1h),3.66–3.57(m,2h),3.19(s,3h),2.34(s,3h).13cnmr(126mhz,cdcl3)δ197.3,175.6,142.5,136.9,133.3,131.7,131.6,128.7,128.7,128.3,128.1,127.9,126.8,126.1,123.1,107.7,88.9,83.2,47.9,41.0,30.6,26.3,21.3.hrms(esi)[m+h]+calcdforc27h24no2+,394.1802,found394.1801.
实施例2化合物b的合成
将苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(400mhz,cdcl3)δ8.01(d,j=7.4hz,2h),7.56(t,j=7.3hz,1h),7.46(t,j=7.6hz,2h),7.23–7.13(m,1h),6.85(dd,j=8.4,2.4hz,1h),6.74(d,j=8.5hz,1h),4.10(td,j=7.0,3.6hz,1h),3.80(s,3h),3.78–3.70(m,2h),3.36(dd,j=17.2,6.9hz,1h),3.18(s,3h).13cnmr(101mhz,cdcl3)δ197.8,175.3,156.1,138.2,136.8,133.3,131.6,128.6,128.5,128.3,128.1,127.9,123.0,113.1,111.5,108.2,88.3,88.1,82.9,55.8,48.2,39.7,30.2,26.2.hrms(esi)[m+h]+calcdforc27h24no3+,410.1751,found410.1751.
实施例3化合物c的合成
将苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(500mhz,cdcl3)δ8.01(d,j=7.4hz,2h),7.56(t,j=7.0hz,2h),7.46(t,j=7.4hz,2h),7.32(t,j=7.7hz,1h),7.23–7.17(m,3h),7.13(dd,j=14.8,7.2hz,3h),6.84(d,j=7.7hz,1h),4.11(d,j=6.7hz,1h),3.83–3.66(m,2h),3.37(dd,j=17.2,6.7hz,1h),3.20(s,3h).13cnmr(126mhz,cdcl3)δ197.8,175.7,144.7,136.8,133.3,131.6,128.7,128.5,128.3,128.1,127.9,127.3,124.3,123.0,122.8,107.9,88.3,82.8,47.9,39.7,30.2,26.1.hrms(esi)[m+h]+calcdforc21h22no2+,380.1645,found380.1643.
实施例4化合物d的合成
将苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(500mhz,cdcl3)δ8.01(d,j=7.5hz,2h),7.57(t,j=7.3hz,1h),7.47(t,j=7.4hz,2h),7.32(d,j=7.8hz,1h),7.22(q,j=6.8hz,3h),7.16(d,j=6.9hz,2h),7.03(t,j=8.8hz,1h),6.76(d,j=8.0hz,1h),4.09(s,1h),3.80(dd,j=16.8,7.4hz,2h),3.38(d,j=6.4hz,1h),3.19(s,3h).13cnmr(126mhz,cdcl3)δ197.7,175.3,159.3(d,j=240.5hz),140.7,136.7,133.4,131.6,129.0(d,j=8.4hz),128.7,128.3,128.2,128.1,122.8,114.6(d,j=23.5hz),112.5(d,j=25.0hz),108.3(d,j=8.2hz),87.8,83.0,48.0,39.6,30.1,26.3.19fnmr(376mhz,cdcl3)δ-120.32.hrms(esi)[m+h]+calcdforc21h21no2f+,398.1551,found398.1552.
实施例5化合物e的合成
将苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(400mhz,cdcl3)δ8.05–8.00(m,2h),7.60–7.51(m,2h),7.49–7.43(m,2h),7.31(t,j=7.7hz,1h),7.18(tdd,j=4.7,4.0,1.3hz,3h),7.15–7.07(m,3h),6.85(d,j=7.9hz,1h),4.11(tt,j=6.9,3.3hz,1h),3.84(ddd,j=9.4,7.3,3.7hz,2h),3.76–3.73(m,1h),3.67(dd,j=14.1,7.1hz,1h),3.44–3.36(m,1h),1.20(t,j=7.2hz,3h).13cnmr(101mhz,cdcl3)δ197.9,175.4,143.7,136.8,133.3,131.6,129.1,128.7,128.4,128.3,128.1,127.9,127.7,124.4,122.5,108.0,88.2,83.3,82.7,47.6,39.7,34.6,30.2,12.8.hrms(esi)[m+h]+calcdforc27h24no2+,394.1802,found394.1799.
实施例6化合物f的合成
将苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(500mhz,cdcl3)δ7.85(d,j=7.3hz,2h),7.64(d,j=7.2hz,1h),7.52(t,j=7.2hz,1h),7.42(t,j=7.3hz,2h),7.35(t,j=7.5hz,1h),7.31–7.22(m,5h),7.12(t,j=7.3hz,1h),6.90(d,j=7.6hz,1h),3.88(d,j=9.9hz,1h),3.23(s,3h),2.93–2.76(m,2h),1.62(s,3h).13cnmr(126mhz,cdcl3)δ197.2,178.9,143.3,136.8,133.1,131.8,131.6,128.6,128.5,128.3,128.2,128.0,123.9,123.1,123.0,108.1,88.3,84.4,50.3,39.5,36.1,26.3,22.7.hrms(esi)[m+h]+calcdforc27h24no2+,394.1802,found394.1803.
实施例7化合物g的合成
将4-甲基苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(400mhz,cdcl3)δ8.01(d,j=7.4hz,2h),7.56(t,j=7.3hz,1h),7.46(t,j=7.6hz,2h),7.37(s,1h),7.10(d,j=7.9hz,1h),7.03(dd,j=18.1,8.1hz,4h),6.72(d,j=7.9hz,1h),4.07(td,j=7.0,3.6hz,1h),3.76(dd,j=17.2,7.3hz,1h),3.72(d,j=3.2hz,1h),3.35(dd,j=17.2,6.7hz,1h),3.17(s,3h),2.37(s,3h),2.29(s,3h).13cnmr(126mhz,cdcl3)δ198.0,175.7,142.3,137.9,136.8,133.3,132.2,131.4,128.9,128.6,128.6,128.3,127.4,125.2,120.0,107.5,87.6,82.8,48.0,39.9,30.3,26.1,21.4,21.2.hrms(esi)[m+h]+calcdforc28h26no2+,408.1958,found408.1958.
实施例8化合物h的合成
将4-甲氧基苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(500mhz,cdcl3)δ8.02(d,j=7.6hz,2h),7.56(t,j=7.2hz,1h),7.46(t,j=7.4hz,2h),7.38(s,1h),7.10(t,j=8.8hz,3h),6.73(t,j=6.4hz,3h),4.07(t,j=6.7hz,1h),3.76(s,3h),3.75–3.70(m,2h),3.35(dd,j=17.2,6.7hz,1h),3.18(s,3h),2.37(s,3h).13cnmr(101mhz,cdcl3)δ198.0,175.7,159.3,142.3,136.8,133.3,132.9,132.2,128.6,128.6,128.3,127.4,125.2,115.2,113.7,107.5,86.8,82.6,55.2,48.0,39.9,30.3,26.1,21.3.hrms(esi)[m+h]+calcdforc28h26no3+,424.1907,found424.1906.
实施例9化合物i的合成
将4-氯苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(400mhz,cdcl3)δ8.04–7.95(m,2h),7.57(t,j=7.4hz,1h),7.47(t,j=7.6hz,2h),7.35(s,1h),7.20–7.17(m,1h),7.17–7.15(m,1h),7.12(d,j=7.9hz,1h),7.08–7.06(m,1h),7.05(d,j=1.8hz,1h),6.73(d,j=7.9hz,1h),4.07(td,j=7.0,3.6hz,1h),3.78(dd,j=17.3,7.2hz,1h),3.72–3.68(m,1h),3.39(dd,j=17.3,6.8hz,1h),3.18(s,3h),2.37(s,3h).13cnmr(101mhz,cdcl3)δ197.8,175.6,142.3,136.7,133.9,133.4,132.8,132.3,128.7,128.7,128.4,128.3,127.2,125.1,121.5,107.6,89.5,81.7,47.9,39.8,30.2,26.1,21.3.hrms(esi)[m+h]+calcdforc27h23no2cl+,428.1412,found428.1412.
实施例10化合物j的合成
将4-甲氧甲酰基苯丙炔醛(0.3mmol)、对氯苯胺(0.18mmol)、烯丙基氯化钯二聚体(0.015mmol)、(s)-6,6'-双(三苯基硅基)螺环二酚磷酸酯(0.03mmol)、
1hnmr(500mhz,cdcl3)δ8.02(d,j=7.4hz,2h),7.88(d,j=7.8hz,2h),7.58(t,j=7.2hz,1h),7.48(t,j=7.3hz,2h),7.36(s,1h),7.19(d,j=7.8hz,2h),7.13(d,j=7.8hz,1h),6.74(d,j=7.8hz,1h),4.14–4.07(m,1h),3.89(s,3h),3.81(dd,j=17.4,7.1hz,1h),3.73(s,1h),3.42(dd,j=17.3,6.6hz,1h),3.19(s,3h),2.38(s,3h).13cnmr(126mhz,cdcl3)δ197.7,175.5,166.5,142.3,136.7,133.4,132.3,131.5,129.3,129.3,128.8,128.7,128.3,127.8,127.2,125.1,107.6,91.8,82.1,52.2,47.8,39.7,30.3,26.2,21.2.hrms(esi)[m+h]+calcdforc29h26no4+,452.1856,found452.1857.
实施例11化合物k的合成
在干燥的10ml磨口反应管中加入产物a(117.9mg,0.30mmol)、缓慢搅拌下加入三氟乙酸(1.5ml),加入完毕后,在常温下继续反应30分钟。反应结束后,加入饱和碳酸氢钠溶液,待不再冒出气泡后,加入乙酸乙酯萃取,分液合并有机相后用无水硫酸钠干燥,减压浓缩有机溶液后,加2ml二氯甲烷,加入0.5ml三乙胺,搅拌6小时后浓缩溶液通过柱层析分离纯化得到纯产物k,产率83%,ee值90%。该产物的单晶衍射图如图1所示,1hnmr示意图如图24所示,其13cnmr示意图如图25所示。
1hnmr(500mhz,cdcl3)δ7.81–7.76(m,2h),7.53–7.49(m,1h),7.42–7.35(m,1h),7.23(dd,j=16.2,8.5hz,2h),6.82(dd,j=10.4,7.8hz,2h),6.29(d,j=6.2hz,1h),3.38(ddd,j=11.4,6.2,3.4hz,1h),3.28(s,3h),3.00(dd,j=15.9,3.4hz,1h),2.48(dd,j=15.9,11.4hz,1h),1.55(s,3h).13cnmr(101mhz,cdcl3)δ198.1,180.9,142.3,138.1,137.4,136.8,133.1,130.6,129.9,129.4,128.5,128.4,128.4,128.1,128.1,127.8,118.4,107.4,46.1,39.1,36.9,26.6,21.9.hrms(esi)[m+h]+calcdforc27h24no2+,384.1802,found384.1806.
实施例12化合物l的合成
在干燥的10ml磨口反应管中加入产物f(39.3mg,0.10mmol)、缓慢搅拌下加入三氟乙酸(0.5ml),加入完毕后,在常温下继续反应30分钟。反应结束后,加入饱和碳酸氢钠溶液,待不再冒出气泡后,加入乙酸乙酯萃取,分液合并有机相后用无水硫酸钠干燥,减压浓缩有机溶液后通过柱层析分离纯化(ea:pe=1:10~1:5)得到纯产物,收率为88%,dr值大于20:1,ee值为90%。
在干燥的10ml磨口反应管中加入上述产物(39.3mg,0.10mmol)、吡啶(31.6mg,0.4mmol)、盐酸羟胺(27.8mg,0.4mmol)和乙醇(2ml),常温下反应过夜。反应结束后,分别用食盐水和3n盐酸洗涤,再经无水硫酸钠干燥后减压浓缩有机溶液后通过柱层析分离纯化(ea:pe=1:2~1:1)得到纯的肟化产物l,产率为90%,e:z值>20:1,ee值为90%。该产物的单晶衍射图如图2示,1hnmr示意图如图26所示,其13cnmr示意图如图27所示。
1hnmr(500mhz,cdcl3)δ7.99(s,1h),7.50–7.46(m,2h),7.37–7.28(m,6h),7.23(t,j=7.8hz,1h),7.17–7.13(m,2h),6.80(dd,j=14.8,7.8hz,2h),5.80(d,j=6.1hz,1h),3.28(dd,j=10.9,6.5hz,1h),3.25(s,3h),2.82(dd,j=12.4,4.9hz,1h),2.42(t,j=11.8hz,1h),1.49(s,3h).13cnmr(126mhz,cdcl3)δ180.9,142.6,138.2,137.8,135.6,130.8,129.5,129.1,128.8,128.6,128.4,128.3,128.0,127.7,126.4,118.3,107.4,46.5,37.8,26.5,24.9,21.7.hrms(esi)[m+h]+calcdforc27h25n2o2+,409.1911,found409.1908.(chiralia3,λ=254nm,n-hexane/2-propanol=80/20,flowrate=1.0ml/min),tr=8.171min,15.581min(major).
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!