一种多官能团环己烯醛化合物的合成及其应用的制作方法
本发明涉及药物应用技术领域,特别涉及一种多官能团环己烯醛化合物的合成及其应用。
背景技术:
官能团对有机物的性质起决定作用,-x、-oh、-cho、-cooh、-no2、-so3h、-nh2、rco-等各种官能团就决定了有机物中的卤代烃、醇或酚、醛、羧酸、硝基化合物或亚硝酸酯、磺酸类有机物、胺类、酰胺类的化学性质,而多官能团环己烯醛类化合物具有非常活跃的反应特性和优良的物理性能,分子结构中存在着十分活泼的环氧基团,能生成一系列重要的有机化合物,这些化合物广泛用于农药、涂料、树脂、表面活性剂等领域,因此对其进行研究既有理论价值也有实际价值,近年来由于环己烯环氧化反应的研究发展很快,因此很必要对其合成方法进行综述。
目前多官能团环己烯醛类化合物大多是通过人工设计和有机合成来得到,由于其易于进行结构修饰,并能方便地引入各种功能基团,所以这些化合物具体微观结构的变化多样,在中间体合成等方面有较为广泛的应用,然而在医药领域的应用却非常有限,因此具有良好的开发潜力。
技术实现要素:
本发明的目的在于提供一种多官能团环己烯醛化合物的合成及其应用。
实现本发明的技术解决方案是:一种多官能团环己烯醛化合物(1-(3-叔丁基二苯基硅氧基)丙基-4-羟基-6-甲基2-环己烯-1-醛),其结构如下所示:
上述一种多官能团环己烯醛化合物的制备方法,包括如下步骤:
(1)将钠溶于适量乙醇中,待钠溶解完,依次加入乙酰乙酸乙酯和巴豆酸甲酯,待反应完全后,经过萃取,得到前体化合物3。
(2)将所述前体化合物3溶于适量乙腈中,分别加入碳酸铯和brch2ch2ch2otbdps,萃取干燥后得到前体化合物4。
(3)将所述前体化合物4溶于适量乙醚中,慢慢加入四氢铝锂(lialh4),反应完全后加入盐酸酸化,将萃取干燥后的产物溶于甲醇,与七水三氯化铈(cecl3.7h2o)和硼氢化钠(nabh4)反应完全后得到前体化合物5。
(4)将所述前体化合物5溶于二氯甲烷中,加入imid,tbscl溶于二氯甲烷中,待反应完全有固体生成后,经过淬灭、萃取、干燥、浓缩后得到前体化合物6.
(5)将所述前体化合物6溶于二氯甲烷中,依次加入nahco3和dmp搅拌,待反应完全后经过淬灭、萃取、干燥、浓缩后,将所得到的产物溶于thf中,随后加入hcl搅拌,待反应完全后淬灭、萃取、干燥、浓缩后得到前体化合物x。
附图说明
图1为前体化合物3的(1hnmr,400mhz,溶剂cdcl3)的核磁共振图谱。
图2为前体化合物4的(1hnmr,400mhz,溶剂cdcl3)的核磁共振图谱。
图3为前体化合物5的(1hnmr,400mhz,溶剂cdcl3)的核磁共振图谱。
图4为前体化合物6的(1hnmr,400mhz,溶剂cdcl3)的核磁共振图谱。
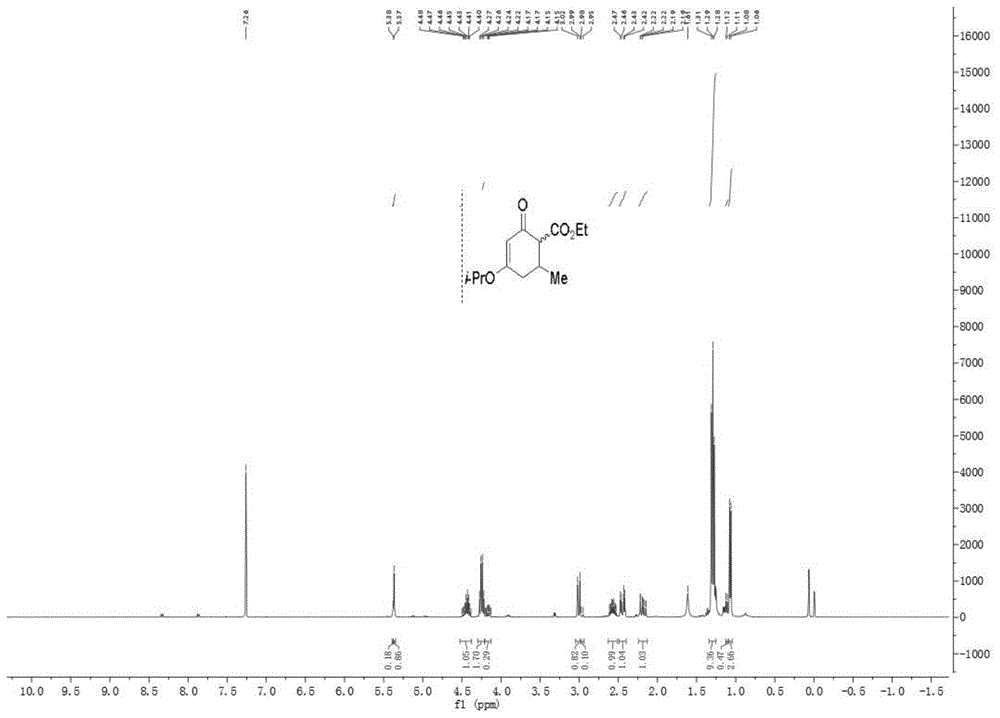
图5为化合物x的(1hnmr,400mhz,溶剂cdcl3)的核磁共振图谱。
图6为化合物x抗菌作用形态学透射电镜图。
具体实施方式
结合以下具体实施案例,对本发明做进一步的详细说明,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明及权利要求书中所涉及的保护范围。
本发明一种多官能团环己烯醛化合物的合成及其应用,通过多轮合成,制造出一种多官能团环己烯醛化合物(1-(3-叔丁基二苯基硅氧基)丙基-4-羟基-6-甲基2-环己烯-1-醛),其结构如下所示:
该化合物的具体合成步骤及方法为:
(1)将钠溶于适量乙醇中,待钠溶解完,依次加入乙酰乙酸乙酯1和巴豆酸甲酯2,升温回流,有白色固体生成。待反应完全后,旋干,加入水至固体完全溶解,用乙酸乙酯萃取三次,保留水相,使用浓盐酸化,然后用乙酸乙酯萃取,无水硫酸钠干燥,得到白色固体,加入异丙醇和樟脑磺酸(csa)以及甲苯,回流分水,反应完全,有机相经浓缩和柱色谱分离,得到前体化合物3,其反应化学式如下所示:
(2)将前体化合物3溶于适量乙腈中,分别加入碳酸铯(cs2co3),brch2ch2ch2otbdps,加热至反应结束,加入水到体系中,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱色谱分离,得到前体化合物4,其反应化学式如下所示:
(3)将前体化合物4溶于适量乙醚中,降至0℃,慢慢加入四氢铝锂(lialh4),反应完全后加入盐酸酸化,用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到的粗产物溶于甲醇,冷却至0℃,依次加入七水三氯化铈(cecl3.7h2o)和硼氢化钠(nabh4),反应完全后用饱和氯化铵淬灭,二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩,柱色谱分离得到前体化合物5,其反应化学式如下所示:
(4)将前体化合物5(21.0g,47.87mmol)溶于200ml的二氯甲烷中,冷却至0℃,依次加入imid(4.9g,71.80mmol),将tbscl(7.2g,47.87mmol)溶于50ml的二氯甲烷中,在0℃的条件下,使用恒压滴液漏斗慢慢滴加至上述溶液中,约0.5h-1h。待有固体生成,tlc检测反应完全,磷钼酸显色。饱和碳酸氢钠淬灭,二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩,得到前体化合物6(17.8g,67.5%),其反应化学式如下所示:
(5)将前体化合物6(20.0g,36.17mmol)溶于300ml的二氯甲烷中,在室温条件下,依次加入nahco3(6.1g,72.34mmol),dmp(18.4g,43.40mmol),搅拌5h,tlc检测反应完全,磷钼酸显色,饱和碳酸氢钠和硫代硫酸钠淬灭,二氯甲烷萃取(100mlx3),有机相用无水硫酸钠干燥,浓缩,直接下一步。
在0℃条件下,将上述粗产物溶于60ml的thf中,随后加入2mhcl(10ml),搅拌6h,进行tlc检测,磷钼酸显色。待反应完全,用饱和碳酸氢钠淬灭,乙酸乙酯(80mlx3)萃取,无水硫酸钠干燥,浓缩,柱色谱,得到化合物x(9.8g,62%),其反应化学式如下所示:
利用质谱(ms)、核磁共振波谱(nmr)、红外吸收光谱(ir)和紫外吸收光谱(uv)等有机波谱,对上述合成的系列新型香豆素类化合物进行分子量、结构和纯度等鉴定。
其鉴定结果为:
前体化合物3
1hnmr(400mhz,溶剂cdcl3):δ1.08(d,j=6.5hz,2.6h),1.12(d,j=6.8hz,0.47h),1.29(t,j=7.0hz,9h),2.15-2.22(m,1h),2.47(dd,j=4.8,17.4hz,1h),2.53-2.61(m,1h),2.98(d,j=11.3hz,0.11h),3.02(d,j=11.4hz,0.82h),4.13-4.19(m,0.29h),4.28(q,j=7.1hz,1.7h),4.38(m,1h),5.37(s,0.86h),5.38(s,0.14h)ppm,其核磁共振图谱可见图1。
前体化合物4
1hnmr(400mhz,溶剂cdcl3):δ1.04-1.05(m,12h),1.23(t,j=7.1hz,3h),1.32(t,j=6.1hz,6h),1.34-1.50(m,2h),1.96(dt,j=4.5,12.2hz,1h),2.21-2.35(m,3h),2.51-2.58(m,1h),3.59-3.74(m,2h),4.09-4.16(m,2h),4.50(septet,j=5.9hz,1h),5.41(s,1h),7.35-7.41(m,6h),7.65-7.88(m,4h)ppm,其核磁共振图谱可见图2。
前体化合物5
1hnmr(400mhz,溶剂cdcl3):δ0.96(d,j=6.6hz,3h),1.03(s,9h),1.23-1.26(m,1h),1.35-1.46(m,3h),1.64-1.73(m,4h),1.81-1.86(m,1h),3.33(d,j=11.2hz,1h),3.56-3.62(m,3h),4.23(d,j=8.1hz,1h),5.34(dd,j=1.9,10.4hz,1h),5.91(d,j=10.1hz,1h),7.35-7.41(m,6h),7.65(d,j=6.8hz,4h),其核磁共振图谱可见图3。
前体化合物6
1hnmr(400mhz,溶剂cdcl3):δ0.10(s,3h),0.12(s,3h),0.93(s,9h),0.98(d,j=6.1hz,3h),1.11(s,9h),1.27-1.32(m,1h),1.39-1.49(m,4h),1.71-1.73(m,3h),5.31(dd,j=1.9,10.2hz,1h),5.85(dd,j=1.6,10.2hz,1h),7.37-7.46(m,6h),7.69(d,j=6.4hz,4h)ppm,其核磁共振图谱可见图4。
化合物x
1hnmr(400mhz,溶剂cdcl3):δ1.01(d,j=6.9hz,3h),1.05(s,9h),1.38-1.53(m,2h),1.60-1.69(m,2h),1.88-1.95(m,1h),2.00-2.05(m,1h),3.71(dt,j=1.3,6.3hz,2h),4.35(t,j=6.3hz,1h),5.31(dd,j=2.0,10.1hz,1h),7.36-7.43(m,6h),7.67(m,4h,),9.66(s,1h)ppm,其核磁共振图谱可见图5。
实施例:合成化合物的最小抑菌浓度(mic)的测定
试验中所用菌株来自美国模式培养物研究所(americantypeculturecollection,atcc),采用液体稀释法测定。按照256、128、64、32、16、8、4、2、1、0.5、0.25、μg/ml的浓度梯度配制化合物。摇菌至a630nm=0.5~0.6,稀释至0.5麦氏比浊标准,然后1:300稀释,取50μl的菌液加入不同浓度的化合物,37℃孵育12~16h。同时设置m-h肉汤对照和单纯细菌对照。在每管中加入10g/l(1%)氯化三苯四氮唑(ttc)10μl。37℃孵育3h后有细菌生长管呈红色,不显红色的最低药物浓度为化合物对检测菌的mic。
测定结果如表1所示。
化合物抗菌作用的形态学研究:
取经过化合物1-5处理,培养旺盛生长的金黄色葡萄球菌(atcc29213)2小时后。
(1)收集菌体,取培养菌液,8000rpm离心3~5min,弃上清,倒入2.5%戊二醛固定液。
(2)固定脱水按常规方法,2.5%戊二醛2~4h,磷酸缓冲液清洗3次,1%锇酸4~6h,缓冲液清洗3次,乙醇梯度脱水,乙酸异戊酯置换2次,20min/次。
(3)临界点干燥:普通定性滤纸裁成35mm×18mm的纸条,将长边35mm平均分成3份,对折成小纸包,将离心浓缩后的菌液滴入小纸包,放入临界点干燥器样品室,进行co2临界点干燥。
(4)离子溅射金:剪开滤纸包,将干燥后粉末状纯菌体倒入平皿,轻摇尽量分散菌体。碳导电胶带一面粘在1/4盖玻片上,另一面倒扣轻压在菌体粉末上,翻正后用镊子将菌体轻轻刮薄铺平,离子溅射金后,即可进行扫描电镜观察,化合物x抗菌作用形态学透射电镜图可见图6。
对实验结果分析发现,将没有经过化合物处理的细菌作为空白对照组(图a),以及化合物1(图b)、2(图c)、9(图d)、10(图e)、11(图f)孵育金黄色葡萄球菌2小时后,和对照组相比,化合物1和2处理后无明显变化,而化合物9,10,11孵育后细菌拟核区致密物质有明显改变,提示化合物9,10,11可以通过抑制敏感菌的遗传物质影响其生长和繁殖。
表1
- 还没有人留言评论。精彩留言会获得点赞!