一种铜催化三组分反应合成三取代烯基硼酸酯的方法
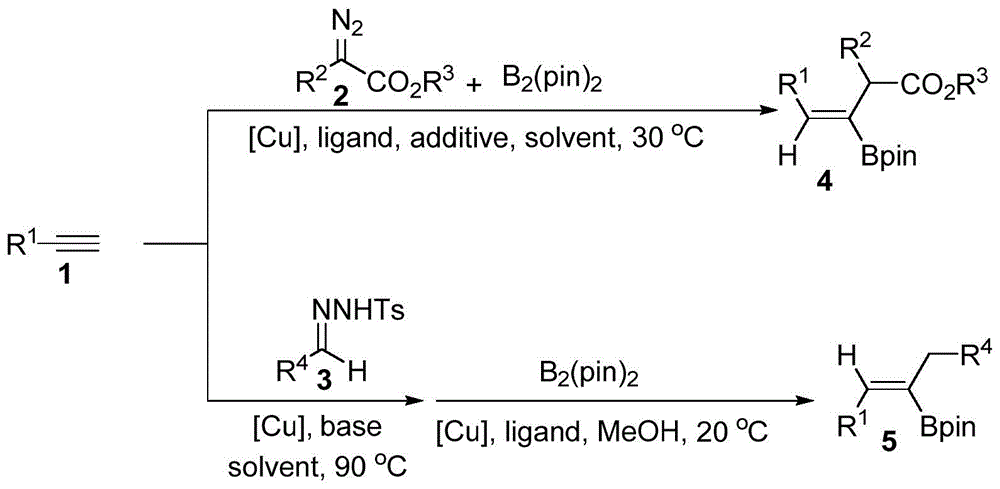
本发明属于有机合成领域,具体涉及一种使用铜催化的端炔、双联频哪醇硼酸酯与重氮化合物或磺酰腙一锅法合成烯基硼酸酯的方法。
背景技术:
有机硼酸酯是一类具有重要合成应用价值的有机试剂,被广泛应用于交叉偶联反应中。烯基硼酸酯作为最常见的复杂烯烃衍生物的合成子,能够用来合成含烯基的目标分子。目前合成烯基硼酸酯的主要方法是通过金属催化烯烃、联烯或炔烃与硼试剂的硼化反应制备(moure,a.l.,etal.j.am.chem.soc.2012,134,7219.reid,w.b.;,etal.org.lett.2018,20,6832.magre,m.,etal.angew.chem.int.ed.2019,58,7025.brzozowska,a.,etal.org.lett.2020,22,3765.guo,w.-h.,etal.acscatal.2019,9,38.hu,y.,etal.angew.chem.int.ed.2019,58,15813.)。
通过n-磺酰腙与硼试剂的硼化也是合成有机硼化合物的有效方法(li,h.;wang,l.;zhang,y.;wang,j.angew.chem.,int.ed.2012,51,2943.li,h.;shangguan,x.;zhang,z.;huang,s.;zhang,y.;wang,j.org.lett.2014,16,448.),尽管合成烯基硼酸酯的方法很多,但三组分一锅反应在同一个炔端碳双官能团化的得到三取代烯基硼酸酯的合成还未实现。
技术实现要素:
本发明公开了一种铜催化的端炔(1)、双联频哪醇硼酸酯(b2(pin)2)与重氮化合物(2)或n-磺酰腙(3)一锅法合成三取代烯基硼酸酯(4)或(5)的方法,反应经历的过程首先是铜催化端炔与重氮化合物或n-磺酰腙偶联生成活泼的联烯中间体,之后原位与双联频哪醇硼酸酯在铜催化下高立体选择性硼化得到三取代烯基硼酸酯。本发明具体的反应通式如下:
一种铜催化三组分反应合成烯基硼酸酯的方法,具体按照下述步骤进行:
当采用重氮化合物作为卡宾前体时:氩气保护下,端炔(1)、双联频哪醇硼酸酯(b2(pin)2)、重氮化合物(2)、铜催化剂、配体及添加剂在溶剂中30℃反应18小时得到(z)-三取代烯基硼酸酯(4)。
当采用n-磺酰腙作为卡宾前体:氩气保护下,端炔(1)、n-磺酰腙(3)、铜催化剂、碱在溶剂中90℃反应3小时,冷却至室温后加入铜催化剂、配体、双联频哪醇硼酸酯(b2(pin)2)及甲醇在20℃继续反应5小时得到(e)-式三取代烯基硼酸酯(5)。
其中,铜催化剂可以为碘化亚铜,氯化亚铜,三氟甲磺酸铜,六氟磷酸四(乙腈)铜,四氟硼酸四(乙腈)铜,最优的催化剂为碘化亚铜。
反应的溶剂可以为n,n-二甲基甲酰胺,n,n-二甲基乙酰胺,二甲亚砜,二氯甲烷,甲苯,四氢呋喃,乙腈,1,4-二氧六环等。当使用重氮化合物为原料反应时最优溶剂为n,n-二甲基甲酰胺,当使用n-磺酰腙为原料反应时最优溶剂为1,4-二氧六环。
配体可以为二齿氮配体(各种联吡啶,菲啰啉)、膦配体(三苯基膦,三环己基膦,binap,tbuxphos,xantphos)等。当使用重氮化合物为原料反应时最优的配体为2,2’-联吡啶,当使用n-磺酰腙为原料反应时最优的配体为4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(xantphos)。
使用重氮化合物为原料时端炔(1)、b2(pin)2、重氮化合物(2)、铜催化剂、联吡啶配体的摩尔比范围为1:1.5~3:1~1.5:0.08:0.08~0.1,其中,最佳摩尔比为1:3:1.5:0.08:0.1;
使用n-磺酰腙为原料时端炔(1)、b2(pin)2、n-磺酰腙(3)、铜催化剂、膦配体、碱、甲醇的摩尔比范围为:1:1~3:1~2.2:0.2:0.1:1.5~3.5:1.5~4,最佳摩尔比为1:3:2.2:0.2:0.1:3.5:4;
反应的浓度范围为0.01~0.5mol/l;最优反应浓度为:0.05mol/l。
反应的添加剂为分子筛(
使用n-磺酰腙为原料时反应的碱可以为叔丁醇钠,叔丁醇钾,叔丁醇锂,碳酸铯;最优的碱为叔丁醇钠。
合成的三取代烯基硼酸酯为(z)-三取代烯基硼酸酯,结构式如式4所示,或(e)-三取代烯基硼酸酯,结构式如式5所示
其中,三取代烯基硼酸酯(4和5)的结构中:r1可以是各种取代苯环(4-omec6h4,4-mec6h4,4-fc6h4,4-brc6h4,4-phc6h4,3-omec6h4,3-clc6h4等)、萘环、胡椒环、芳杂环、烷基(丁基,苄基)、环烷基(环丙基,环乙基),烯基;
r2可以是各种取代苯环(4-cf3c6h4,4-omec6h4,4-mec6h4,4-fc6h4,4-brc6h4,4-phc6h4,2-fc6h4,4-nhbocc6h4等)、萘环、胡椒环、噻吩等;
r3可以是甲酯、乙酯、烯丙酯;
r4可以是各种烷基(环己基,异丙基)、苯基等。
有益效果
本发明公开了一种铜催化的多组分反应一锅法立体选择性合成三取代烯基硼酸酯的方法,本发明方法克服了末端炔烃端位直接双官能团化合成三取代烯基硼酸酯的立体选择性问题,为直接一步合成多取代烯基硼酸酯提供一种新方法。
本发明方法的优点有:反应原料简单易得,三组分一锅法反应操作简单、产率高、底物适用范围广、区域和立体选择性好;可以通过底物和配体等条件的改变,可调控选择性的生成(z)-三取代烯基硼酸酯或者(e)-三取代烯基硼酸酯。
附图说明
图1为本发明的具体反应工艺过程图;
图2为实施例1得到的4a的1hnmr(核磁氢谱)图;
图3为实施例1得到的4a的13cnmr(核磁碳谱)图;
图4为实施例1得到的4a的hrms(高分辨质谱)图。
具体实施方式
下面将通过具体实施例对本发明做进一步说明,本发明并不局限于以下的实施例:
实施例1:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例2:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例3:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(101.5mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例4:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、pcy3(5.6mg)、
实施例5:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例6:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、
实施例7:4a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cu(otf)2(5.8mg)、
实施例8:4b的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例9:4c的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例10:4d的合成:
氩气保护下,向反应管中加入1a(20.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例11:4e的合成:
氩气保护下,向反应管中加入1b(26.4mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例12:4f的合成:
氩气保护下,向反应管中加入1c(21.6mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例13:4g的合成:
氩气保护下,向反应管中加入1d(13.2mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例14:4h的合成:
氩气保护下,向反应管中加入1e(41.8mg)、b2(pin)2(152.3mg)、cui(3.1mg)、2,2’-联吡啶(3.1mg)、
实施例15:5a的合成:
氩气保护下,向反应管中加入1a(20.4mg)、3a(123.2mg)、cui(3.8mg)、叔丁醇钠(67.2mg)和1,4-二氧六环(4ml),反应混合物在90℃加热模块上搅拌3小时。将反应液冷却至室温并在氩气氛围下,加入cui(3.8mg)、xantphos(11.57mg)、b2(pin)2(152.3mg)和甲醇(25.6mg),反应混合物在20℃搅拌5小时。反应液减压蒸除溶剂后通过柱层析纯化,用乙酸乙酯:石油醚(1:100)淋洗得到无色油状产物5a(41.8mg,64%,e:z>19:1)。1hnmr(400mhz,cdcl3)δ7.34-7.32(m,2h),7.26-7.23(m,2h),7.20-7.16(m,1h),6.83(s,1h),2.19(d,j=7.0hz,2h),1.79-1.63(m,5h),1.48-1.37(m,1h),1.25(s,12h),1.19-1.10(m,3h),0.94-0.85(m,2h).13cnmr(75mhz,cdcl3)δ141.0,139.1,128.2,127.9,127.0,83.6,46.3,38.2,33.5,26.8,26.6,24.9.hrms(esi)m/z:[m+na]+calcdforc21h31bnao2349.2309;found349.2322.
实施例16:5b的合成:
氩气保护下,向反应管中加入1a(20.4mg)、3b(105.7mg)、cui(3.8mg)、叔丁醇钠(67.2mg)和1,4-二氧六环(4ml),反应混合物在90℃加热模块上搅拌3小时。将反应液冷却至室温并在氩气氛围下,加入cui(3.8mg)、xantphos(11.6mg)、b2(pin)2(152.3mg)和甲醇(25.6mg),反应混合物在20℃搅拌5小时。反应液减压蒸除溶剂后通过柱层析纯化,用乙酸乙酯:石油醚(1:100)淋洗得到无色油状产物5b(40.1mg,70%,e:z>19:1)。1hnmr(400mhz,cdcl3)δ7.36-7.31(m,2h),7.28-7.23(m,2h),7.21-7.16(m,1h),6.84(s,1h),2.18(dd,j=7.1,0.8hz,2h),1.82-1.71(m,1h),1.25(s,12h),0.93(d,j=6.6hz,6h).13cnmr(75mhz,cdcl3)δ141.1,139.2,128.2,128.0,127.0,83.6,47.9,28.6,24.9,22.8.hrms(esi)m/z:[m+na]+calcdforc18h27bnao2309.1996;found309.2019.
实施例17:5c的合成:
氩气保护下,向反应管中加入1a(20.4mg)、3c(132.9mg)、cui(3.8mg)、叔丁醇钠(67.2mg)和1,4-二氧六环(4ml),反应混合物在90℃加热模块上搅拌3小时。将反应液冷却至室温并在氩气氛围下,加入cui(3.8mg)、xantphos(11.6mg)、b2(pin)2(152.3mg)和甲醇(25.6mg),反应混合物在20℃搅拌5小时。反应液减压蒸除溶剂后通过柱层析纯化,用乙酸乙酯:石油醚(1:100)淋洗得到无色油状产物5c(25.8mg,37%,e:z=5:1)。1hnmr(400mhz,cdcl3)δ7.36-7.26(m,4h),7.25-7.23(m,1h),7.22-7.14(m,5h),6.89(s,1h),2.69-2.64(m,2h),2.35(t,j=7.4hz,2h),1.83(s,2h),1.25(s,12h).13cnmr(75mhz,cdcl3)δ142.8,140.4,139.1,128.6,128.4,128.2,128.0,127.1,125.7,83.7,37.8,35.6,31.5,24.9.hrms(esi)m/z:[m+h]+calcdforc23h30bo2349.2333;found349.2342.
实施例18:5d的合成:
氩气保护下,向反应管中加入1a(20.4mg)、3d(120.6mg)、cui(3.8mg)、叔丁醇钠(67.2mg)和1,4-二氧六环(4ml),反应混合物在90℃加热模块上搅拌3小时。将反应液冷却至室温并在氩气氛围下,加入cui(3.8mg)、xantphos(11.6mg)、b2(pin)2(152.3mg)和甲醇(25.6mg),反应混合物在20℃搅拌5小时。反应液减压蒸除溶剂后通过柱层析纯化,用乙酸乙酯:石油醚(1:100)淋洗得到无色油状产物5d(29.5mg,46%,e:z>19:1)。1hnmr(300mhz,cdcl3)δ7.41(s,1h),7.34-7.30(m,4h),7.25-7.12(m,6h),3.77(s,2h),1.19(s,12h).13cnmr(75mhz,cdcl3)δ143.2,141.6,137.6,129.1,128.6,128.31,128.27,127.5,125.7,83.7,35.2,24.7.hrms(esi)m/z:[m+na]+calcdforc21h25bnao2343.1840;found343.1846.
实施例19:5d的合成:
氩气保护下,向反应管中加入1a(20.4mg)、3d(120.6mg)、cui(3.8mg)、叔丁醇钾(67.2mg)和1,4-二氧六环(4ml),反应混合物在90℃加热模块上搅拌3小时。将反应液冷却至室温并在氩气氛围下,加入cui(3.8mg)、tbuxphos(8.5mg)、b2(pin)2(152.3mg)和甲醇(25.6mg),反应混合物在20℃搅拌5小时。反应液减压蒸除溶剂后通过柱层析纯化,用乙酸乙酯:石油醚(1:100)淋洗得到无色油状产物5d(21.1mg,33%,e:z=18:1)。
实施例20:5d的合成:
氩气保护下,向反应管中加入1a(20.4mg)、3d(120.6mg)、cui(3.8mg)、叔丁醇钠(67.2mg)和1,4-二氧六环(4ml),反应混合物在90℃加热模块上搅拌3小时。将反应液冷却至室温并在氩气氛围下,加入cui(3.8mg)、binap(12.4mg)、b2(pin)2(152.3mg)和甲醇(25.6mg),反应混合物在20℃搅拌5小时。反应液减压蒸除溶剂后通过柱层析纯化,用乙酸乙酯:石油醚(1:100)淋洗得到无色油状产物5d(23.7mg,37%,e:z=2:1)。
- 还没有人留言评论。精彩留言会获得点赞!