茂金属化合物、烯烃聚合用催化剂组分和烯烃聚合用催化剂、和生产烯烃聚合物的方法与流程
茂金属化合物、烯烃聚合用催化剂组分和烯烃聚合用催化剂、和生产烯烃聚合物的方法
1.本技术是申请日为2017年3月21日、申请号为2017800152735、发明名称为“茂金属化合物、包含其的烯烃聚合用催化剂组分和烯烃聚合用催化剂、和使用烯烃聚合用催化剂生产烯烃聚合物的方法”的中国专利申请的分案申请。
技术领域
2.本发明涉及茂金属化合物、包含其的烯烃聚合用催化剂组分和烯烃聚合用催化剂、和使用烯烃聚合用催化剂生产烯烃聚合物的方法。更具体地,本发明涉及在环戊二烯并噻吩的3位具有芳基取代基或杂芳基取代基并且具有桥接型(bridged)(环戊二烯)
‑
(环戊二烯并噻吩)作为基本骨架的茂金属化合物,包含所述茂金属化合物的烯烃聚合用催化剂组分和烯烃聚合用催化剂,和使用烯烃聚合用催化剂生产烯烃聚合物(特别地,乙烯系聚合物)的方法。
背景技术:
3.作为用于改善在成形加工性方面通常较差的茂金属系聚乙烯的成形加工性的方法,已知将高压法低密度聚乙烯共混至茂金属系聚乙烯中的方法和使用特定的茂金属通过聚合反应将长支链(long
‑
chain branches)引入至聚乙烯中的方法。由于前者需要共混步骤,因此生产成本变高。此外,所得共混物在成形加工性方面优异,但是作为茂金属系聚乙烯的优势的机械强度降低。另一方面,已知如下方法:使用桥接双茚基化合物(例如,参见专利文献1)或几何约束半茂金属(参见专利文献2)作为后者用于引入长支链的特定的茂金属。
4.此外,专利文献3报道了,当使用其中环戊二烯基和茚基与碳桥接的不对称型茂金属和甲基铝氧烷通过溶液聚合来进行乙烯的均聚时,可以生产具有支链的聚乙烯。
5.此外,专利文献4报道了如下催化剂体系,其用于使用其中环戊二烯基和茚基与硅桥接的不对称型茂金属中的在茚基的2、4和7位具有甲基的茂金属和改性粘土化合物生产各自用作大分子单体的乙烯聚合物和乙烯/丁烯共聚物。
6.此外,本发明人已经在专利文献5中提出了,包含其中环戊二烯基和茚基与桥接基团桥接的不对称型茂金属中的在环戊二烯基上除了该桥接基团以外不具有取代基并且在茚基的3位具有氢或特定取代基的特定不对称型茂金属作为必需组分的烯烃聚合用负载型催化剂,另外,提出了使用烯烃聚合用负载型催化剂生产具有改善的成形加工性的乙烯系聚合物的方法。
7.现有技术文献
8.专利文献
9.专利文献1:jp
‑
a
‑
08
‑
048711
10.专利文献2:jp
‑
a
‑
07
‑
500622
11.专利文献3:jp
‑
a
‑
05
‑
043619
12.专利文献4:jp
‑
a
‑
2008
‑
050278
13.专利文献5:jp
‑
a
‑
2011
‑
137146
技术实现要素:
14.发明要解决的问题
15.然而,根据专利文献1和2,所得聚合物中的末端双键和长支链的数量少,因此成形加工性的改善效果尚不充分。
16.根据专利文献3,支链的长度记载为1至20的碳数,并且支链的长度对于显示作为长支链的成形加工性的改善效果而言太短。
17.根据专利文献4,聚合物的末端双键的数量少并且没有仅通过催化剂形成长支链的记载。
18.此外,根据专利文献5,由于获得了伸长粘度的应变硬化度(degree of strain hardening)大的乙烯系聚合物,因而观察到了与常规的长支链型聚乙烯相比成形加工性的改进,但是长支链的支化指数尚未达到高压法低密度聚乙烯的长支链的支化指数,因此要求长支链结构的进一步改进。
19.在这样的情况下,为了改善茂金属系聚乙烯的成形加工性,在早期要求开发具有在其中引入的充分数量和长度的长支链的茂金属系聚乙烯的生产方法。
20.考虑到上述常规技术中的问题,为了改善茂金属系聚乙烯的成形加工性,本发明的目的在于提供能够生产具有在其中引入的充分数量和长度的长支链的乙烯系聚合物的茂金属化合物,包含该茂金属化合物的烯烃聚合用催化剂组分和烯烃聚合用催化剂,并且进一步提供使用烯烃聚合用催化剂生产烯烃聚合物(特别地,乙烯系聚合物)的方法。
21.另外,在本发明中,聚乙烯为乙烯均聚物和乙烯与后面提及的烯烃的共聚物的总称,并且可以改述为乙烯系聚合物。
22.用于解决问题的方案
23.作为为了解决上述问题而广泛研究的结果,本发明人已经发现,当在环戊二烯并噻吩的3位具有芳基取代基或杂芳基取代基并且具有桥接型(环戊二烯)
‑
(环戊二烯并噻吩)作为基本骨架的新型茂金属化合物用作烯烃聚合用催化剂组分、并且使用通过将茂金属化合物与和该茂金属化合物反应以形成阳离子性茂金属化合物的化合物和细颗粒载体组合而获得的催化剂组合物时,可以生产具有充分数量和长度的长支链的茂金属系聚乙烯。基于这些发现,他们完成了本发明。
24.即,本发明提供:
25.[1]一种由以下通式(1)表示的茂金属化合物:
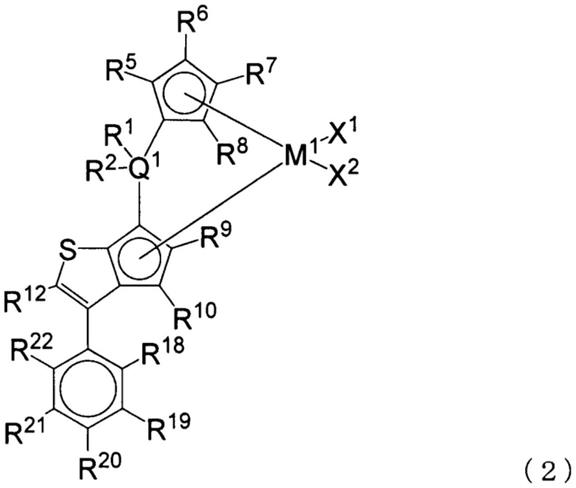
[0026][0027]
在式(1)中,m1表示ti、zr和hf中的任意的过渡金属;x1和x2各自独立地表示氢原子、卤素、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代的氨基、或碳数为1至20的烷氧基;q1和q2各自独立地表示碳原子、硅原子或锗原子;r1、r2、r3和r4各自独立地表示氢原子或碳数为1至10的烃基,并且可以与结合至其的q1和q2中的至少一个一起形成环;m为0或1,并且在m为0的情况下,q1直接结合至包含r9和r
10
的共轭五元环;r5、r6、r7、r8、r9、r
10
和r
12
各自独立地表示氢原子、卤素、碳数为1至20的烃基、包含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、含氧的碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基;和r
11
表示由以下通式(1
‑
a)表示的取代或未取代的芳基:
[0028][0029]
在式(1
‑
a)中,y1表示周期表的第14族、第15族或第16族的原子;r
13
、r
14
、r
15
、r
16
和r
17
各自独立地表示氢原子、卤素原子、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代的氨基、碳数为1至20的烷氧基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、或由碳数为1至20的烃基取代的甲硅烷基,并且r
13
、r
14
、r
15
、r
16
和r
17
中相邻取代基可以与结合至其的原子一起形成环;n为0或1,并且在n为0的情况下,在y1上不存在取代基r
13
;p为0或1,并且在p为0的情况下,取代基r
16
和与r
16
结合的碳原子不存在,并且与r
15
结合的碳原子和与r
17
结合的碳原子直接结合。
[0030]
[2]一种由以下通式(2)表示的茂金属化合物:
[0031][0032]
在式(2)中,m1表示ti、zr和hf中的任意的过渡金属;x1和x2各自独立地表示氢原子、卤素、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代的氨基、或碳数为1至20的烷氧基;q1表示碳原子、硅原子或锗原子;r1和r2各自独立地表示氢原子或碳数为1至10的烃基,并且可以与结合至其的q1一起形成环;r5、r6、r7、r8、r9、r
10
和r
12
各自独立地表示氢原子、卤素、碳数为1至20的烃基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、含氧的碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基;r
18
、r
19
、r
20
、r
21
和r
22
各自独立地表示氢原子、卤素原子、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代的氨基、碳数为1至20的烷氧基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、或由碳数为1至20的烃基取代的甲硅烷基,并且r
18
、r
19
、r
20
、r
21
和r
22
中相邻取代基可以与结合至其的原子一起形成环。
[0033]
[3]一种由以下通式(3)表示的茂金属化合物:
[0034][0035]
在式(3)中,m1表示ti、zr和hf中的任意的过渡金属;x1和x2各自独立地表示氢原子、卤素、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代
的氨基、或碳数为1至20的烷氧基;q1表示碳原子、硅原子或锗原子;r1和r2各自独立地表示氢原子或碳数为1至10的烃基,并且可以与结合至其的q1一起形成环;r5、r6、r7、r8、r9、r
10
和r
12
各自独立地表示氢原子、卤素、碳数为1至20的烃基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、含氧的碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基;z1表示氧原子或硫原子;r
23
、r
24
和r
25
各自独立地表示氢原子、卤素、碳数为1至20的烃基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、含氧的碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基,并且r
23
、r
24
和r
25
中相邻取代基可以与结合至其的碳原子一起形成环。
[0036]
[4]根据[1]至[3]任一项所述的茂金属化合物,其中在以上通式(1)、(2)或(3)中q1为硅原子。
[0037]
[5]根据[1]至[4]任一项所述的茂金属化合物,其中在以上通式(1)、(2)或(3)中r9为氢原子。
[0038]
[6]根据[1]至[5]任一项所述的茂金属化合物,其中在以上通式(1)、(2)或(3)中m1为锆或铪。
[0039]
[7]根据[1]至[6]任一项所述的茂金属化合物,其中在以上通式(1)、(2)或(3)中m1为锆。
[0040]
[8]一种烯烃聚合用催化剂组分,其包含根据[1]至[7]任一项所述的茂金属化合物。
[0041]
[9]一种烯烃聚合用催化剂,其包含根据[1]至[7]任一项所述的茂金属化合物。
[0042]
[10]一种烯烃聚合用催化剂,其包含以下必要组分(a)、(b)和(c):
[0043]
组分(a):根据[1]至[7]任一项所述的茂金属化合物,
[0044]
组分(b):与组分(a)的茂金属化合物反应以形成阳离子性茂金属化合物的化合物,和
[0045]
组分(c):细颗粒载体。
[0046]
[11]根据[10]所述的烯烃聚合用催化剂,其中组分(b)为铝氧烷。
[0047]
[12]根据[10]或[11]所述的烯烃聚合用催化剂,其中组分(c)为二氧化硅。
[0048]
[13]根据[10]至[12]任一项所述的烯烃聚合用催化剂,其进一步包含以下组分(d):
[0049]
组分(d):有机铝化合物。
[0050]
[14]一种烯烃系聚合物的生产方法,其包括使用根据[9]至[13]任一项所述的烯烃聚合用催化剂使烯烃聚合。
[0051]
[15]根据[14]所述的烯烃系聚合物的生产方法,其中烯烃至少包含乙烯。
[0052]
[16]根据[15]所述的烯烃系聚合物的生产方法,其中烯烃系聚合物为乙烯系聚合物。
[0053]
发明的效果
[0054]
本发明的茂金属化合物为在环戊二烯并噻吩的3位具有芳基取代基或杂芳基取代基并且具有桥接型(环戊二烯)
‑
(环戊二烯并噻吩)作为基本骨架的新型茂金属化合物,并且可以生产具有充分数量和长度的长支链的茂金属系聚乙烯。另外,通过使用本发明的茂金属化合物作为烯烃聚合用催化剂组分,可以获得具有其中引入的充分数量和长度的长支
链并且具有进一步改善的成形加工性的烯烃系聚合物(特别地,乙烯系聚合物)。
附图说明
[0055]
图1为说明gpc中色谱图的基线和区间(section)的图。
[0056]
图2为示出由gpc
‑
vis测量计算的支化指数(g’)与分子量(m)之间的关系的图。
具体实施方式
[0057]
以下将详细地描述本发明的茂金属化合物、包含其的烯烃聚合用催化剂组分和烯烃聚合用催化剂、和使用烯烃聚合用催化剂生产烯烃聚合物的方法。
[0058]
1.茂金属化合物
[0059]
本发明的茂金属化合物的特征在于:由以下通式(1)表示的环戊二烯基环和环戊二烯并噻吩基环桥接,并且进一步地,环戊二烯并噻吩环的3位(r
11
)表示取代或未取代的芳基或杂芳基(下文中,有时称为“特定的芳基”)。
[0060][0061]
在本发明的茂金属化合物中,由于环戊二烯基环和环戊二烯并噻吩基环桥接并且在环戊二烯并噻吩环的3位(r
11
)处存在特定的芳基,因而通过β
‑
氢消除的大分子单体(末端乙烯基聚合物)的形成和大分子单体的共聚二者均可以通过使用本发明的茂金属化合物作为烯烃聚合用催化剂组分来进行。因此,当使用包含本发明的茂金属化合物的催化剂时,引入了充分数量和长度的长支链并且可以获得具有进一步改善的成形加工性的烯烃系聚合物(特别地,乙烯系聚合物)。
[0062]
在本发明的茂金属化合物中,对于形成长支链最重要的结构为:环戊二烯并噻吩环用作基本骨架并且在其3位(r
11
)存在特定的芳基。与茚环同样地,环戊二烯并噻吩环可以作为一价阴离子与金属结合。然而,在结构方面的差异在于,茚环由与金属直接结合的五元环结构和与其稠合的六元环结构组成,但是环戊二烯并噻吩环由其中含硫五元环结构与和金属直接结合的五元环结构稠合的结构组成。因此,推测取代至环戊二烯并噻吩环的3位(r
11
)的特定的芳基与芳基取代至茚环上的相应的位置(4位)的情况会在聚合反应中的位阻效应方面产生差异。具体地,在环戊二烯并噻吩环的情况下,由于取代至3位的特定的芳基与茚环的情况相比配置在空间上远离金属的位置,因而推测促进了β
‑
氢消除反应,由此促进了对聚合物中的长支链结构的形成重要的大分子单体(末端乙烯基聚合物)的形成。
[0063]
在通式(1)中,m1表示ti、zr和hf中的任意的过渡金属,优选zr或hf,并且更优选
zr。
[0064]
在通式(1)中,x1和x2各自独立地表示氢原子、卤素、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代的氨基、或碳数为1至20的烷氧基。
[0065]
由x1和x2表示的卤素包括氯原子、溴原子和碘原子等。由x1和x2表示的碳数为1至20的烃基包括烷基和芳基等,并且其实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、新戊基、环戊基、正己基、环己基、苯基、苄基和萘基等。
[0066]
由x1和x2表示的含氧的碳数为1至20的烃基包括具有醚键、羰基、酯键或杂芳基等的烃基,并且其实例包括甲氧基甲基、乙氧基甲基、正丙氧基甲基、异丙氧基甲基、正丁氧基甲基、异丁氧基甲基、叔丁氧基甲基、甲氧基乙基、乙氧基乙基、乙酰基、1
‑
氧代丙基、1
‑
氧代
‑
正丁基、2
‑
甲基
‑1‑
氧代丙基、2,2
‑
二甲基
‑1‑
氧代
‑
丙基、苯基乙酰基、二苯基乙酰基、苯甲酰基、2
‑
甲氧基苯基、3
‑
甲氧基苯基、4
‑
甲氧基苯基、2
‑
呋喃基、2
‑
四氢呋喃基、和2
‑
(5
‑
甲基)呋喃基等。含氮的碳数为1至20的烃基包括具有氨基、亚氨基、氰基(nitrile group)、吡啶基、吡咯基、咪唑基、吡唑基或吲哚基等的烃基,并且其实例包括二甲基氨基甲基、二乙基氨基甲基、二
‑
异丙基氨基甲基、双(二甲基氨基)甲基、双(二
‑
异丙基氨基)甲基、(二甲基氨基)(苯基)甲基、甲基亚氨基、乙基亚氨基、1
‑
(甲基亚氨基)乙基、1
‑
(苯基亚氨基)乙基、和1
‑
[(苯基甲基)亚氨基]乙基等。
[0067]
由x1和x2表示的由碳数为1至20的烃基取代的氨基的实例包括二甲基氨基、二乙基氨基、二
‑
正丙基氨基、二
‑
异丙基氨基、二
‑
正丁基氨基、二
‑
异丁基氨基、二
‑
叔丁基氨基、和二苯基氨基等。
[0068]
由x1和x2表示的碳数为1至20的烷氧基的实例包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基和苯氧基等。
[0069]
优选的x1和x2包括卤素、碳数为1至4的烷基、碳数为1至6的烷氧基、和由碳数为1至6的烃基取代的氨基。其中,可以提及氯原子、溴原子、甲基、正丁基、异丁基、甲氧基、乙氧基、异丙氧基、正丁氧基、苯氧基、二甲基氨基和二
‑
异丙基氨基。其中,氯原子、甲基和二甲基氨基是特别优选的。
[0070]
在通式(1)中,q1和q2各自独立地表示碳原子、硅原子或锗原子,优选碳原子或硅原子,并且更优选硅原子。
[0071]
在通式(1)中,r1、r2、r3和r4各自独立地表示氢原子或碳数为1至10的烃基,并且可以与结合至其的q1和q2中的至少一个一起形成环。m为0或1,并且在m为0的情况下,q1直接结合至包含r9和r
10
的共轭五元环。
[0072]
由r1、r2、r3和r4表示的碳数为1至10的烃基包括烷基和芳基等,并且其实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、新戊基、环戊基、正己基、环己基和苯基等。
[0073]
此外,作为r1和r2与结合至其的q1一起形成环或者r3和r4与结合至其的q2一起形成环的情况,可以提及环丁叉基、环戊叉基、环己叉基、硅杂环丁基、硅杂环戊基和硅杂环己基等。此外,作为r1、r2、r3和r4与结合至其的q1和q2一起形成环的情况,可以提及亚环己基等。
[0074]
优选的r1、r2、r3和r4在q1和/或q2为碳原子的情况下包括氢原子、甲基、乙基、苯基、亚乙基和环丁叉基,并且在q1和/或q2为硅原子的情况下包括甲基、乙基、苯基和硅杂环丁基。
[0075]
在通式(1)中,r5、r6、r7、r8、r9、r
10
和r
12
各自独立地表示氢原子、卤素、碳数为1至20的烃基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、含氧的碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基。
[0076]
在通式(1)中,当r5、r6、r7和r8中的至少一个具有如卤素、碳数为1至20的烃基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、含氧的碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基中的取代基时,特别是聚合活性提高,因此该情形是优选的。包含在r5、r6、r7和r8中的氢原子以外的上述取代基的数量可以为1以上且4以下的任一者,但是优选为2以上且4以下。
[0077]
作为由r5、r6、r7、r8、r9、r
10
和r
12
各自表示的除了氢原子以外的取代基的具体实例,关于卤素、碳数为1至20的烃基或含氧的碳数为1至20的烃基,可以提及与前述x1和x2的描述中所示的基团相同的那些。
[0078]
含1至6的硅数的碳数为1至18的含硅烃基的实例包括双(三甲基甲硅烷基)甲基和双(叔丁基二甲基甲硅烷基)甲基等,并且碳数为1至20的含卤素烃基的实例包括溴甲基、氯甲基、三氟甲基、2
‑
氯乙基、2
‑
溴乙基、2
‑
溴丙基、3
‑
溴丙基、2
‑
溴环丙基、2,3
‑
二溴环戊基、2
‑
溴
‑3‑
碘环戊基、2,3
‑
二溴环己基、2
‑
氯
‑3‑
碘环己基、2
‑
氯苯基、4
‑
氯苯基、2,3,4,5,6
‑
五氟苯基、和4
‑
三氟甲基苯基等。
[0079]
此外,由碳数为1至20的烃基取代的甲硅烷基包括三烷基甲硅烷基、二烷基单芳基甲硅烷基、单烷基二芳基甲硅烷基和三芳基甲硅烷基等,并且其实例包括三甲基甲硅烷基、三叔丁基甲硅烷基、二叔丁基甲基甲硅烷基、叔丁基二甲基甲硅烷基、三苯基甲硅烷基、二苯基甲基甲硅烷基、和苯基二甲基甲硅烷基等。
[0080]
优选的r5、r6、r7、r8、r9、r
10
和r
12
为氢原子、碳数为1至20的烃基、或由碳数为1至20的烃基取代的甲硅烷基,更优选的是氢原子、碳数为1至10的烃基、或由碳数为1至18的烃基取代的甲硅烷基,并且甚至进一步优选的是氢原子、碳数为1至6的烃基、或由碳数为1至6的烃基取代的甲硅烷基。
[0081]
碳数为1至10的烃基的实例包括碳数为1至10的烷基、可以由碳数6至10的烷基取代的苯基、和萘基。由碳数为1至18的烃基取代的甲硅烷基的优选实例包括三甲基甲硅烷基、乙基二甲基甲硅烷基、正丙基二甲基甲硅烷基、异丙基二甲基甲硅烷基、正丁基二甲基甲硅烷基、异丁基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、三乙基甲硅烷基、叔丁基二乙基甲硅烷基、三异丙基甲硅烷基、二甲基苯基甲硅烷基、甲基二苯基甲硅烷基、和叔丁基二苯基甲硅烷基。
[0082]
此外,碳数为1至6的烃基的优选实例包括碳数为1至6的烷基,并且碳数为1至6的烷基的优选实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、新戊基、环戊基、正己基和环己基。
[0083]
特别优选的r5、r6、r7、r8、r9、r
10
和r
12
为氢原子、碳数为1至6的烷基、或由碳数为1至6的烃基取代的甲硅烷基。作为除了氢原子以外的取代基,更优选的是甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、三甲基甲硅烷基、三乙基甲硅烷基、或叔丁基二甲基甲硅烷基,并且进一步优选的是甲基、叔丁基或三甲基甲硅烷基。
[0084]
取代基r
11
表示具有由以上通式(1
‑
a)表示的结构的取代或未取代的芳基。通式(1
‑
a)中的y1优选为碳原子、氮原子、氧原子和硫原子中的任一种,并且进一步优选碳原子、氧
原子和硫原子中的任一种。优选的具有由以上通式(1
‑
a)表示的结构的未取代的芳基包括苯基、呋喃基和噻吩基等。
[0085]
在通式(1
‑
a)中,r
13
、r
14
、r
15
、r
16
和r
17
各自独立地表示氢原子、卤素原子、碳数为1至20的烃基、含氧或氮的碳数为1至20的烃基、由碳数为1至20的烃基取代的氨基、碳数为1至20的烷氧基、含1至6的硅数的碳数为1至18的含硅烃基、碳数为1至20的含卤素烃基、或由碳数为1至20的烃基取代的甲硅烷基,并且r
13
、r
14
、r
15
、r
16
和r
17
中相邻取代基可以与结合至其的原子一起形成一个以上的环。
[0086]
作为由r
13
、r
14
、r
15
、r
16
和r
17
各自表示的除了氢原子以外的取代基的具体实例,可以提及与前述x1和x2以及r5、r6、r7、r8、r9、r
10
和r
12
的描述中示出的基团相同的那些。
[0087]
取代基r
11
中的具有由以上通式(1
‑
a)表示的结构的取代或未取代的芳基具体包括苯基、4
‑
甲基苯基、4
‑
异丙基苯基、4
‑
叔丁基苯基、4
‑
联苯基、2,6
‑
二甲基苯基、2,5
‑
二甲基苯基、3,5
‑
二甲基苯基、3,5
‑
异丙基苯基、3,5
‑
叔丁基苯基、2,4,6
‑
三甲基苯基、2,3,5,6
‑
四甲基苯基、2,3,4,5,6
‑
五甲基苯基、4
‑
三甲基甲硅烷基苯基、4
‑
(叔丁基二甲基甲硅烷基)苯基、3,5
‑
双三甲基甲硅烷基苯基、4
‑
氟苯基、4
‑
氯苯基、4
‑
溴苯基、4
‑
三氟甲基苯基、3,5
‑
二氟苯基、3,5
‑
二氯苯基、2,4,6
‑
三氟苯基、3,4,5
‑
三氟苯基、2,4,6
‑
三氯苯基、2,3,5,6
‑
四氟苯基、2,3,4,5,6
‑
五氟苯基、4
‑
甲氧基苯基、4
‑
乙氧基苯基、4
‑
异丙氧基苯基、4
‑
正丁氧基苯基、4
‑
苯氧基苯基、3,5
‑
二甲氧基苯基、2
‑
呋喃基、2
‑
(5
‑
甲基)呋喃基、2
‑
(5
‑
正丙基)呋喃基、2
‑
(5
‑
异丙基)呋喃基、2
‑
(5
‑
正丁基)呋喃基、2
‑
(5
‑
异丁基)呋喃基、2
‑
(5
‑
叔丁基)呋喃基、2
‑
(5
‑
三甲基甲硅烷基)呋喃基、2
‑
(5
‑
三乙基甲硅烷基)呋喃基、2
‑
(5
‑
苯基)呋喃基、2
‑
(5
‑
甲苯基)呋喃基、2
‑
(5
‑
氟苯基)呋喃基、2
‑
(5
‑
氯苯基)呋喃基、2
‑
(4,5
‑
二甲基)呋喃基、2
‑
苯并呋喃基、2
‑
噻吩基、2
‑
(5
‑
甲基)噻吩基、2
‑
(5
‑
叔丁基)噻吩基、2
‑
(5
‑
三甲基甲硅烷基)噻吩基、和2
‑
(4,5
‑
二甲基)噻吩基等。
[0088]
此外,r
13
、r
14
、r
15
、r
16
和r
17
中相邻取代基可以与结合至其的原子一起形成一个以上的芳香族环或脂肪族环。
[0089]
形成的芳香族环或脂肪族环的优选实例包括1
‑
萘基、2
‑
萘基、1
‑
蒽基、2
‑
蒽基、9
‑
蒽基、1
‑
菲基、2
‑
菲基、3
‑
菲基、4
‑
菲基、9
‑
菲基、5
‑
1,2,3,4
‑
四氢萘基、6
‑
1,2,3,4
‑
四氢萘基和9
‑
1,2,3,4,5,6,7,8
‑
八氢蒽基。其中,更优选1
‑
萘基、2
‑
萘基和9
‑
蒽基,并且进一步优选1
‑
萘基和2
‑
萘基。
[0090]
此外,在通式(1)中,m为0或1,并且在m为0的情况下,q1直接结合至包含r9和r
10
的共轭五元环。
[0091]
本发明的茂金属化合物优选为由以下通式(2)表示的茂金属化合物。
[0092][0093]
在由以上通式(2)表示的茂金属化合物中,作为m1、x1、x2、q1、r1、r2、r5、r6、r7、r8、r9、r
10
和r
12
,可以选择与由前述通式(1)表示的茂金属化合物的描述中所示的原子和基团相同的结构。此外,作为r
18
、r
19
、r
20
、r
21
和r
22
,可以选择与由前述通式(1)表示的茂金属化合物的描述中所示的r
13
、r
14
、r
15
、r
16
和r
17
的原子和基团相同的结构。
[0094]
此外,本发明的茂金属化合物优选为由与以上通式(2)相似的以下通式(3)表示的茂金属化合物。
[0095][0096]
在由以上通式(3)表示的茂金属化合物中,作为m1、x1、x2、q1、r1、r2、r5、r6、r7、r8、r9、r
10
和r
12
,可以选择与由前述通式(1)表示的茂金属化合物的描述中所示的原子和基团相同的结构。此外,作为r
23
、r
24
和r
25
,可以选择与由前述通式(1)表示的茂金属化合物的描述中所示的r
13
、r
14
、r
15
、r
16
和r
17
的原子和基团相同的结构。z1表示氧原子或硫原子。
[0097]
作为本发明的茂金属化合物,在由以上通式(1)表示的化合物中,可以提及由以下通式(4)表示的化合物作为优选化合物。
[0098][0099]
在由以上通式(4)表示的茂金属化合物中,作为m1、x1、x2、q、r1、r2、r9、r
11
和r
12
,可以选择与由前述通式(1)表示的茂金属化合物的描述中所示的原子和基团相同的构造。
[0100]
本发明的茂金属化合物的具体实例在以下通式(1’)和表1至表5中示出,但是不限于此。
[0101]
另外,在以下表中,缩写如下。
[0102]
me:甲基,et:乙基,pr:丙基,bu:丁基,ph:苯基,cp:环戊二烯基
[0103][0104]
[表1]
[0105]
表1
[0106]
编号mx1,x2r1r2qr9r5r6r7r8cpr
10
r
11
r
12
1zrclme2simecphphme2zrclme2simecph4
‑
me
‑
phme3zrclme2simecph4
‑
i
pr
‑
phme4zrclme2simecph4
‑
t
bu
‑
phme5zrclme2simecph4
‑
ph
‑
phme6zrclme2simecph4
‑
meo
‑
phme7zrclme2simecph4
‑
pho
‑
phme8zrclme2simecph4
‑
f
‑
phme9zrclme2simecph4
‑
cl
‑
phme10zrclme2simecph4
‑
cf3‑
phme11zrclme2simecph2,6
‑
me2‑
phme12zrclme2simecph2,5
‑
me2‑
phme13zrclme2simecph3,5
‑
me2‑
phme14zrclme2simecph3,5
‑
i
pr2‑
phme
15zrclme2simecph3,5
‑
t
bu2‑
phme16zrclme2simecph3,5
‑
(meo)2‑
phme17zrclme2simecph3,5
‑
f2‑
phme18zrclme2simecph3,5
‑
cl2‑
phme19zrclme2simecph2,4,6
‑
me3‑
phme20zrclme2simecph2,4,6
‑
f3‑
phme21zrclme2simecph3,4,5
‑
f3‑
phme22zrclme2simecph2,3,5,6
‑
me4‑
phme23zrclme2simecph2,3,5,6
‑
f4‑
phme24zrclme2simecph2,3,4,5,6
‑
me5‑
phme25zrclme2simecph2,3,4,5,6
‑
f5‑
phme26zrbrme2simecphphme27zrmeme2simecphphme28zrnme2me2simecphphme29zroch3me2simecphphme30zroetme2simecphphme31zro
t
bume2simecphphme32zrophme2simecphphme33zrclme2sietcphphme34zrclme2si
n
prcphphme35zrclme2si
i
prcphphme36zrclme2si
n
bucphphme37zrclme2si
i
bucphphme38zrclme2si
t
bucphphme39zrclme2siphcphphme40zrclme2si1
‑
萘基cphphme
[0107]
[表2]
[0108]
表2
[0109][0110]
[表3]
[0111]
表3
[0112][0113]
[表4]
[0114]
表4
[0115][0116]
[表5]
[0117]
表5
[0118][0119]
此外,作为优选的化合物,可以提及其中上述化合物的锆被钛或铪代替的化合物等。
[0120]
2.茂金属化合物的合成方法
[0121]
本发明的茂金属化合物可以根据取代基或结合方式通过任意的方法来合成。代表的合成路线的一个实例在以下示出。
[0122][0123]
在以上合成路线中,通过使1和苯基溴化镁在镍催化剂的存在下进行偶联反应来获得2。在2与甲基丙烯酸反应之后,将所得3用氢化铝锂还原并且使用对甲苯磺酸进行进一步的脱水,从而获得氢环戊二烯环戊二烯并噻吩5。在将5用1当量的正丁基锂等阴离子化之后,使该阴离子与过量的二甲基二氯硅烷反应并且通过蒸馏除去未反应的二甲基二氯硅烷从而获得6。使所得6与环戊二烯基钠反应从而获得7。在将7用2当量的正丁基锂等二阴离子化之后,使该双阴离子(dianion)与四氯化锆反应从而获得8。
[0124]
关于其中取代基引入至在环戊二烯并噻吩环的3位的苯基的茂金属化合物的合成,可以通过使用相应的取代的原料来实现该合成。通过使用例如4
‑
甲基苯基溴化镁、4
‑
异丙基苯基溴化镁或4
‑
叔丁基苯基溴化镁等相应的格氏试剂代替苯基溴化镁,可以在环戊二烯并噻吩环的3位引入相应的取代基。
[0125]
此外,通过使用例如二乙基二氯硅烷或二苯基二氯硅烷等相应的试剂代替二甲基二氯硅烷来与5反应,可以引入相应的桥接基团结构。
[0126]
此外,通过使用例如叔丁基环戊二烯、1,3
‑
二甲基环戊二烯或1
‑
甲基
‑3‑
叔丁基环戊二烯的阴离子等相应的取代的环戊二烯的阴离子来代替环戊二烯基,可以合成其中相应的取代基引入至环戊二烯的配合物。
[0127]
此外,关于其中在环戊二烯并噻吩环的4位引入各种取代基的茂金属化合物的合成,其中在环戊二烯并噻吩环的4位引入各种取代基的5可以通过以下来合成:使3与对应于各种取代基的格氏试剂或有机锂试剂反应而不是与氢化铝锂反应、并且用对甲苯磺酸进行脱水反应。
[0128]
3.烯烃聚合用催化剂
[0129]
(1)各组分
[0130]
本发明的茂金属化合物形成烯烃聚合用催化剂组分并且该催化剂组分可以用于
烯烃聚合用催化剂。
[0131]
本发明的烯烃聚合用催化剂除了前述本发明的茂金属化合物以外还可以包含已知的组分,但是优选包含以下组分(a)、(b)和(c)。
[0132]
组分(a):本发明的茂金属化合物
[0133]
组分(b):与组分(a)的茂金属化合物反应以形成阳离子性茂金属化合物的化合物,和
[0134]
组分(c):细颗粒载体。
[0135]
(2)组分(a)
[0136]
本发明的烯烃聚合用催化剂使用由任意的前述通式(1)至(3)表示的茂金属化合物作为必要组分(a),并且也可以使用其一种或两种以上化合物。
[0137]
(3)组分(b)
[0138]
本发明的烯烃聚合用催化剂除了以上组分(a)以外,优选包含作为组分(b)的与组分(a)的茂金属化合物反应以形成阳离子性茂金属化合物的化合物。
[0139]
对组分(b)不特别限定,只要其为与组分(a)反应以形成阳离子性茂金属化合物的化合物即可,并且可以使用已知的组分,但是例如可以提及有机铝氧化合物、硼烷化合物和硼酸盐化合物(borate compound)等。
[0140]
当有机铝氧化合物用作组分(b)时,所得乙烯系聚合物的应变硬化度(λmax)变大和/或作为高分子量组分含量的量度的mz/mw(其中mz表示通过gpc测量的z平均分子量和mw表示由gpc测量的重均分子量)增大,并且由此进一步改善了成形性,从而优选该用途。
[0141]
当硼烷化合物或硼酸盐化合物用作组分(b)时,聚合活性和共聚性变高,从而改善了具有长支链的乙烯系聚合物的生产性。
[0142]
此外,作为组分(b),也可以使用以上有机铝氧化合物与以上硼烷化合物或硼酸盐化合物的混合物。此外,硼烷化合物和硼酸盐化合物也可以以其两种以上的组合使用。
[0143]
以下将进一步详细地描述各化合物。
[0144]
(i)有机铝氧化合物
[0145]
有机铝氧化合物在分子中具有al
‑
o
‑
al键,并且键的个数通常在1至100、优选1至50的范围内。这样的有机铝氧化合物通常为通过使有机铝化合物与水反应获得的产物。
[0146]
在有机铝氧化合物中,通过使烷基铝与水反应获得的化合物通常称为铝氧烷并且可以适当地用作组分(b)。另外,在铝氧烷中,甲基铝氧烷(包括实质上由甲基铝氧烷(mao)组成的那些)特别适合作为有机铝氧化合物。
[0147]
另外,作为有机铝氧化合物,也可以组合使用两种以上的有机铝氧化合物,并且可以使用溶解或分散在以下将提及的惰性烃溶剂中的有机铝氧化合物的溶液。
[0148]
有机铝与水的反应通常在惰性烃(溶剂)中进行。作为惰性烃,可以使用例如戊烷、己烷、庚烷、环己烷、甲基环己烷、苯、甲苯和二甲苯等脂肪族烃、脂环族烃和芳香族烃,但是优选使用脂肪族烃或芳香族烃。
[0149]
作为要用于制备有机铝氧化合物的有机铝化合物,可以使用任意的由以下通式(i)表示的化合物,但是优选使用三烷基铝。
[0150]
r
at
alx
a3
‑
t
ꢀꢀꢀꢀꢀ
(i)
[0151]
其中r
a
表示碳数为1至18、优选碳数为1至12的烃基,如烷基、烯基、芳基或芳烷基,
x
a
表示氢原子或卤素原子,和t表示1≤t≤3的整数。
[0152]
三烷基铝中的烷基的实例包括甲基、乙基、丙基、异丙基、丁基、异丁基、戊基、己基、辛基、癸基和十二烷基等,但是其中甲基特别优选的。
[0153]
以上有机铝化合物也可以以其一种或两种以上的组合使用。
[0154]
水与有机铝化合物的反应比(水/al的摩尔比)优选为0.25/1至1.2/1,特别优选0.5/1至1/1,并且反应温度通常在
‑
70至100℃、优选
‑
20至20℃的范围内。反应时间通常选自5分钟至24小时、优选10分钟至5小时的范围内。作为反应所需的水,不仅可以利用单纯的水,还可以利用包含在硫酸铜水合物或硫酸铝水合物等中的结晶水和可以在反应体系中生成水的组分。
[0155]
(ii)硼烷化合物
[0156]
此外,要用作组分(b)的硼烷化合物的实例包括三苯基硼烷、三(邻甲苯基)硼烷、三(对甲苯基)硼烷、三(间甲苯基)硼烷、三(邻氟苯基)硼烷、三(对氟苯基)硼烷、三(间氟苯基)硼烷、三(2,5
‑
二氟苯基)硼烷、三(3,5
‑
二氟苯基)硼烷、三(4
‑
三氟甲基苯基)硼烷、三(3,5
‑
二三氟甲基苯基)硼烷、三(2,6
‑
二三氟甲基苯基)硼烷、三(五氟苯基)硼烷、三(全氟萘基)硼烷、三(全氟联苯基)硼烷、三(全氟蒽基)硼烷、和三(全氟联萘基)硼烷等。
[0157]
其中,更优选三(3,5
‑
二三氟甲基苯基)硼烷、三(2,6
‑
二三氟甲基苯基)硼烷、三(五氟苯基)硼烷、三(全氟萘基)硼烷、三(全氟联苯基)硼烷、三(全氟蒽基)硼烷、和三(全氟联萘基)硼烷,并且进一步优选地,示例三(2,6
‑
二三氟甲基苯基)硼烷、三(五氟苯基)硼烷、三(全氟萘基)硼烷和三(全氟联苯基)硼烷作为优选的硼烷化合物。
[0158]
(iii)硼酸盐化合物
[0159]
此外,当具体表示要用作组分(b)的硼酸盐化合物时,第一实例为由以下通式(ii)表示的化合物。
[0160]
[l1‑
h]
+
[br
b
r
c
x
b
x
c
]
—
.....(ii)
[0161]
在式(ii)中,l1为中性路易斯碱,h为氢原子,[l1‑
h]为布朗斯台德酸,如铵、苯胺鎓(anilinium)或鏻。作为铵,可以示例三烷基取代的铵,如三甲基铵、三乙基铵、三丙基铵、三丁基铵和三(正丁基)铵;和二烷基铵,如二(正丙基)铵和二环己基铵。
[0162]
此外,作为苯胺鎓,可以示例n,n
‑
二烷基苯胺鎓,如n,n
‑
二甲基苯胺鎓、n,n
‑
二乙基苯胺鎓和n,n
‑
2,4,6
‑
五甲基苯胺鎓。此外,作为鏻,可以提及例如三苯基鏻、三丁基鏻、三(甲基苯基)鏻、和三(二甲基苯基)鏻等三芳基鏻,以及三烷基鏻。
[0163]
此外,在式(ii)中,r
b
和r
c
各自为包含6至20个、优选6至16个碳原子、彼此相同或不同的芳香族或取代的芳香族烃基,并且可以由桥接基团彼此连接。作为取代的芳香族烃基的取代基,优选的是由甲基、乙基、丙基或异丙基表示的烷基,或卤素原子如氟、氯、溴或碘。此外,x
b
和x
c
各自独立地为氢化物基团、卤化物基团、包含1至20个碳原子的烃基、或其中一个以上的氢原子被卤素原子取代的包含1至20个碳原子的取代的烃基。
[0164]
作为由以上通式(ii)表示的化合物的具体实例,可以示例三丁基铵四(五氟苯基)硼酸盐、三丁基铵四(2,6
‑
二三氟甲基苯基)硼酸盐、三丁基铵四(3,5
‑
二三氟甲基苯基)硼酸盐、三丁基铵四(2,6
‑
二氟苯基)硼酸盐、三丁基铵四(全氟萘基)硼酸盐、二甲基苯胺鎓四(五氟苯基)硼酸盐、二甲基苯胺鎓四(2,6
‑
二三氟甲基苯基)硼酸盐、二甲基苯胺鎓四(3,5
‑
二三氟甲基苯基)硼酸盐、二甲基苯胺鎓四(2,6
‑
二氟苯基)硼酸盐、二甲基苯胺鎓四(全氟
萘基)硼酸盐、三苯基鏻四(五氟苯基)硼酸盐、三苯基鏻四(2,6
‑
二三氟甲基苯基)硼酸盐、三苯基鏻四(3,5
‑
二三氟甲基苯基)硼酸盐、三苯基鏻四(2,6
‑
二氟苯基)硼酸盐、三苯基鏻四(全氟萘基)硼酸盐、三甲基铵四(2,6
‑
二三氟甲基苯基)硼酸盐、三乙基铵四(五氟苯基)硼酸盐、三乙基铵四(2,6
‑
二三氟甲基苯基)硼酸盐、三乙基铵四(全氟萘基)硼酸盐、三丙基铵四(五氟苯基)硼酸盐、三丙基铵四(2,6
‑
二三氟甲基苯基)硼酸盐、三丙基铵四(全氟萘基)硼酸盐、二(1
‑
丙基)铵四(五氟苯基)硼酸盐、和二环己基铵四苯基硼酸盐等。
[0165]
其中,优选的是三丁基铵四(五氟苯基)硼酸盐、三丁基铵四(2,6
‑
二三氟甲基苯基)硼酸盐、三丁基铵四(3,5
‑
二三氟甲基苯基)硼酸盐、三丁基铵四(全氟萘基)硼酸盐、二甲基苯胺鎓四(五氟苯基)硼酸盐、二甲基苯胺鎓四(2,6
‑
二三氟甲基苯基)硼酸盐、二甲基苯胺鎓四(3,5
‑
二三氟甲基苯基)硼酸盐、和二甲基苯胺鎓四(全氟萘基)硼酸盐。
[0166]
进一步,硼酸盐化合物的第二实例由以下通式(iii)表示。
[0167]
[l2]
+
[br
b
r
c
x
b
x
c
]
—
.....(iii)
[0168]
在式(iii)中,作为l2,可以提及碳阳离子、甲基阳离子、乙基阳离子、丙基阳离子、异丙基阳离子、丁基阳离子、异丁基阳离子、叔丁基阳离子、戊基阳离子、托品阳离子(tropinium cation)、苄基阳离子、三苯甲基阳离子、钠阳离子和质子等。进一步,r
b
、r
c
、x
b
和x
c
与以上通式(ii)中的定义相同。
[0169]
作为以上化合物的具体实例,可以示例三苯甲基四苯基硼酸盐、三苯甲基四(邻甲苯基)硼酸盐、三苯甲基四(对甲苯基)硼酸盐、三苯甲基四(间甲苯基)硼酸盐、三苯甲基四(邻氟苯基)硼酸盐、三苯甲基四(对氟苯基)硼酸盐、三苯甲基四(间氟苯基)硼酸盐、三苯甲基四(3,5
‑
二氟苯基)硼酸盐、三苯甲基四(五氟苯基)硼酸盐、三苯甲基四(2,6
‑
二三氟甲基苯基)硼酸盐、三苯甲基四(3,5
‑
二三氟甲基苯基)硼酸盐、三苯甲基四(全氟萘基)硼酸盐、托品四苯基硼酸盐、托品四(邻甲苯基)硼酸盐、托品四(对甲苯基)硼酸盐、托品四(间甲苯基)硼酸盐、托品四(邻氟苯基)硼酸盐、托品四(对氟苯基)硼酸盐、托品四(间氟苯基)硼酸盐、托品四(3,5
‑
二氟苯基)硼酸盐、托品四(五氟苯基)硼酸盐、托品四(2,6
‑
二三氟甲基苯基)硼酸盐、托品四(3,5
‑
二三氟甲基苯基)硼酸盐、托品四(全氟萘基)硼酸盐、nabph4、nab(o
‑
ch3‑
ph)4、nab(p
‑
ch3‑
ph)4、nab(m
‑
ch3‑
ph)4、nab(o
‑
f
‑
ph)4、nab(p
‑
f
‑
ph)4、nab(m
‑
f
‑
ph)4、nab(3,5
‑
f2‑
ph)4、nab(c6f5)4、nab(2,6
‑
(cf3)2‑
ph)4、nab(3,5
‑
(cf3)2‑
ph)4、nab(c
10
f7)4、hbph4·
2二乙醚、hb(3,5
‑
f2‑
ph)4·
2二乙醚、hb(c6f5)4·
2二乙醚、hb(2,6
‑
(cf3)2‑
ph)4·
2二乙醚、hb(3,5
‑
(cf3)2‑
ph)4·
2二乙醚、和hb(c
10
h7)4·
2二乙醚。
[0170]
其中,优选的是三苯甲基四(五氟苯基)硼酸盐、三苯甲基四(2,6
‑
二三氟甲基苯基)硼酸盐、三苯甲基四(3,5
‑
二三氟甲基苯基)硼酸盐、三苯甲基四(全氟萘基)硼酸盐、托品四(五氟苯基)硼酸盐、托品四(2,6
‑
二三氟甲基苯基)硼酸盐、托品四(3,5
‑
二三氟甲基苯基)硼酸盐、托品四(全氟萘基)硼酸盐、nab(c6f5)4、nab(2,6
‑
(cf3)2‑
ph)4、nab(3,5
‑
(cf3)2‑
ph)4、nab(c
10
f7)4、hb(c6f5)4·
2二乙醚、hb(2,6
‑
(cf3)2‑
ph)4·
2二乙醚、hb(3,5
‑
(cf3)2‑
ph)4·
2二乙醚、和hb(c
10
h7)4·
2二乙醚。
[0171]
其中,更优选地,可以提及三苯甲基四(五氟苯基)硼酸盐、三苯甲基四(2,6
‑
二三氟甲基苯基)硼酸盐、托品四(五氟苯基)硼酸盐、托品四(2,6
‑
二氟甲基苯基)硼酸盐、nab(c6f5)4、nab(2,6
‑
(cf3)2‑
ph)4、hb(c6f5)4·
2二乙醚、hb(2,6
‑
(cf3)2‑
ph)4·
2二乙醚、hb(3,5
‑
(cf3)2‑
ph)4·
2二乙醚、和hb(c
10
h7)4·
2二乙醚。
[0172]
(4)组分(c)
[0173]
作为为组分(c)的细颗粒载体,本发明的烯烃聚合用催化剂优选使用无机载体、颗粒状聚合物载体、或其混合物。作为无机载体,可以使用金属、金属氧化物、金属氯化物、金属碳酸盐、含碳物质、或其混合物。
[0174]
作为可以用作无机载体的合适的金属,例如可以提及铁、铝和镍等。
[0175]
进一步,作为金属氧化物,可以提及周期表的第1至14族的元素的单独的氧化物或复合氧化物,并且例如可以示例天然或合成的各种单独的氧化物或复合氧化物如,sio2、al2o3、mgo、cao、b2o3、tio2、zro2、fe2o3、al2o3·
mgo、al2o3·
cao、al2o3·
sio2、al2o3·
mgo
·
cao、al2o3·
mgo
·
sio2、al2o3·
cuo、al2o3·
fe2o3、al2o3·
nio和sio2·
mgo等。这里,上述式不是分子式并且仅表示组成,并且对要用于本发明的复合氧化物的结构和催化剂组分比例不特别限定。要用于本发明的金属氧化物可以为已经吸收了少量的水分的金属氧化物并且也可以为包含少量的杂质的金属氧化物。
[0176]
作为金属氯化物,例如,优选碱金属或碱土类金属的氯化物,并且具体地,特别优选mgcl2和cacl2等。作为金属碳酸盐,优选碱金属或碱土类金属的碳酸盐,并且具体地,可以提及碳酸镁、碳酸钙和碳酸钡等。
[0177]
作为含碳物质,例如,可以提及炭黑和活性炭等。
[0178]
以上无机载体均可以适用于本发明,但是特别地,金属氧化物、二氧化硅或氧化铝等的使用是优选的。
[0179]
优选通常将无机载体在200℃至800℃、优选400℃至600℃下在空气或例如氮气或氩气等惰性气体中煅烧从而将表面羟基的量调节至0.8mmol/g至1.5mmol/g之后使用这些无机载体。对无机载体的性质不特别限定,但是通常优选使用具有以下性质的无机载体:5μm至200μm、优选10μm至150μm的平均粒径,至优选至的平均孔径,150m2/g至1000m2/g、优选200m2/g至700m2/g的比表面积,0.3cm3/g至2.5cm3/g、优选0.5cm3/g至2.0cm3/g的孔容积,和0.20g/cm3至0.50g/cm3、优选0.25g/cm3至0.45g/cm3的表观比重。
[0180]
当然,上述无机载体可以以其原样使用,但是作为预处理,可以在使载体与例如三甲基铝、三乙基铝、三异丁基铝、三己基铝、三丙基铝、三丁基铝、三辛基铝、三癸基铝(tridecylaluminum)、或二异丁基氢化铝等有机铝化合物、或包含al
‑
o
‑
al键的有机铝氧化合物接触之后使用。
[0181]
4.烯烃聚合用催化剂的制备方法
[0182]
对在获得由以下组成的烯烃聚合用催化剂时各组分的接触方法不特别限定:作为为本发明的烯烃系聚合物的生产方法的必要组分的茂金属化合物的组分(a)、与组分(a)反应以形成阳离子性茂金属化合物的组分(b)和作为细颗粒载体的组分(c),并且例如,可以任意地采用以下方法。
[0183]
(i)在使组分(a)与组分(b)彼此接触之后,使组分(c)与其接触。
[0184]
(ii)在使组分(a)与组分(c)彼此接触之后,使组分(b)与其接触。
[0185]
(iii)在使组分(b)与组分(c)彼此接触之后,使组分(a)与其接触。
[0186]
在这些接触方法中,接触方法(i)和(iii)是优选的,并且接触方法(i)是最优选的。在任意的接触方法中,通常采用如下方法:在例如氮气或氩气等惰性气氛中,一般在例如,如苯、甲苯、二甲苯或乙基苯等芳香族烃(通常碳数为6至12)或如庚烷、己烷、癸烷、十二
烷或环己烷等脂肪族或脂环族烃(通常碳数为5至12)等液态惰性烃的存在下,在搅拌或不搅拌的情况下,使各组分彼此接触。该接触优选在通常为
‑
100℃至200℃、优选
‑
50℃至100℃、进一步优选0℃至50℃的温度下进行5分钟至50小时、优选30分钟至24小时、更优选30分钟至12小时。
[0187]
此外,作为在组分(a)、组分(b)和组分(c)接触时使用的溶剂,如上所述,可以使用某些组分可溶或难溶于其中的芳香族烃溶剂和某些组分不溶或难溶于其中的脂肪族或脂环族烃溶剂二者。
[0188]
在各组分的接触反应分阶段进行的情况下,用于前阶段的溶剂等可以在随后阶段的接触反应中直接用作溶剂而不将其除去。可选地,在使用可溶性溶剂的前阶段的接触反应之后,可以将某些组分不溶或难溶于其中的液态惰性烃(例如,如戊烷、己烷、癸烷、十二烷、环己烷、苯、甲苯或二甲苯等脂肪族烃、脂环族烃或芳香族烃)添加至其中,从而回收作为固体物质的期望的产物,或者通过例如干燥等手段一次性除去可溶性溶剂的一部分或全部从而取出作为固体物质的期望的产物,此后,期望的产物的随后阶段的接触反应可以使用任意的上述惰性烃溶剂来进行。在本发明中,各组分的接触反应可以进行多次。
[0189]
在本发明中,对组分(a)、组分(b)和组分(c)的使用比例不特别限定,但是优选在以下范围内。
[0190]
在有机铝氧化合物用作组分(b)的情况下,期望有机铝氧化合物中的铝与作为本发明的茂金属化合物的组分(a)中的过渡金属(m)的原子比(al/m)在通常1至100,000、优选5至1,000、更优选50至200的范围内。
[0191]
此外,在硼烷化合物或硼酸盐化合物用作组分(b)的情况下,期望硼与作为本发明的茂金属化合物的组分(a)中的过渡金属(m)的原子比(b/m)在通常0.01至100、优选0.1至50、更优选0.2至10的范围内。
[0192]
此外,在有机铝氧化合物与硼烷化合物或硼酸盐化合物的混合物用作组分(b)的情况下,对于混合物中的各化合物,期望选择分别与上述相同的相对于作为本发明的茂金属化合物的组分(a)中的过渡金属(m)的al和b的使用比例。
[0193]
相对于组分(a)中的过渡金属(m)0.0001至5mmol、优选相对于0.001至0.5mmol、更优选相对于0.01至0.1mmol,要使用的作为细颗粒载体的组分(c)的量为1g。
[0194]
使组分(a)、组分(b)何组分(c)通过任意的接触方法(1)至(3)彼此接触,此后,通过除去溶剂,可以获得烯烃聚合用催化剂作为固体催化剂。期望溶剂的除去在常压下或在减压下在0至200℃、优选在20至150℃下进行1分钟至50小时、优选10分钟至10小时。
[0195]
另外,本发明的烯烃聚合用催化剂也可以通过以下方法获得。
[0196]
(iv)使组分(a)和组分(c)彼此接触并且除去溶剂从而形成固体催化剂组分,使其在聚合条件下与作为有机铝氧化合物、硼烷化合物、硼酸盐化合物、或其混合物的组分(b)接触。
[0197]
(v)使作为有机铝氧化合物、硼烷化合物、硼酸盐化合物、或其混合物的组分(b)与组分(c)接触并且除去溶剂从而形成固体催化剂组分,使其在聚合条件下与组分(a)接触。
[0198]
另外,在以上接触方法(iv)和(v)的情况下,作为组分比、接触条件和溶剂除去条件,可以使用与上述相同的条件。
[0199]
此外,作为用作组分(b)和组分(c)二者的组分,也可以使用层状硅酸盐。层状硅酸
盐为具有其中由离子键等构成的面以弱的键合力平行地堆叠的晶体结构的硅酸盐化合物。大部分的层状硅酸盐主要作为粘土矿物的主要组分而天然地产出,但是这些层状硅酸盐不特别限于天然存在的层状硅酸盐并且可以为人工合成物。
[0200]
其中,例如蒙脱石、锌蒙脱石、贝得石、绿脱石、皂石、锂皂石、硅镁石、膨胀土和带云母等蒙脱石族、蛭石族和云母族是优选的。
[0201]
一般地,天然产物通常为非离子交换性(非溶胀性)的,并且在这种情况下,为了具有优选的离子交换性(或溶胀性),优选进行用于赋予离子交换性(或溶胀性)的处理。在这样的处理中,特别优选包括以下化学处理。这里,作为化学处理,可以使用其中除去附着至表面的杂质的表面处理和影响层状硅酸盐的晶体结构和/或化学组成的处理二者。具体地,可以提及(i)使用盐酸或硫酸等进行的酸处理,(ii)使用naoh、koh或nh3等进行的碱处理,(iii)使用由包含选自周期表的第2族至第14族的至少一种原子的阳离子和选自由卤素原子和源自无机酸的阴离子组成的组中的至少一种阴离子组成的盐的盐处理,和(iv)用例如醇、烃化合物、甲酰胺或苯胺等有机物的处理等。这些处理可以单独进行或以其两种以上的组合进行。
[0202]
关于层状硅酸盐,在任何步骤之前、期间或之后的任何时间,可以通过粉碎、造粒、分选(sizing)或分级等来控制颗粒性质。方法可以是任何合目的的方法。特别地,关于造粒法,例如,可以提及喷雾造粒法、滚动造粒法、压缩造粒法、搅拌造粒法、压块法、压实法、挤压造粒法、流化床造粒法、乳化造粒法和液中造粒法等。其中,特别优选的造粒法为喷雾造粒法、滚动造粒法和压缩造粒法。
[0203]
当然,上述层状硅酸盐可以以其原样使用,但是层状硅酸盐可以与例如三甲基铝、三乙基铝、三异丁基铝、三丙基铝、三丁基铝、三己基铝、三辛基铝、三癸基铝或二异丁基氢化铝等有机铝化合物,或者包含al
‑
o
‑
al键的有机铝氧化合物组合使用。
[0204]
为了在层状硅酸盐上负载作为本发明的烯烃系聚合物的生产方法的必要组分的组分(a),可以使组分(a)与层状硅酸盐彼此接触,或者可以使组分(a)、有机铝化合物和层状硅酸盐彼此接触。
[0205]
对各组分的接触方法不特别限定,并且例如,可以任意地采用以下方法。
[0206]
(vi)在使组分(a)与有机铝化合物彼此接触之后,使所得物与层状硅酸盐载体接触。
[0207]
(vii)在使组分(a)与层状硅酸盐载体彼此接触之后,使所得物与有机铝化合物接触。
[0208]
(viii)在使有机铝化合物与层状硅酸盐载体彼此接触之后,使所得物与组分(a)接触。
[0209]
在这些接触方法中,优选接触方法(iv)和(viii)。即使在任意的接触方法中,仍通常采用如下方法:在例如氮气或氩气等惰性气氛中,一般地在例如,如苯、甲苯、二甲苯或乙基苯等芳香族烃(通常碳数为6至12)或如庚烷、己烷、癸烷、十二烷或环己烷等脂肪族或脂环族烃(通常碳数为5至12)等液态惰性烃的存在下,在搅拌或不搅拌的情况下,使各组分彼此接触。
[0210]
对组分(a)、有机铝化合物和层状硅酸盐载体的使用比例不特别限定,但是优选在以下范围内。组分(a)的负载量相对于1g的层状硅酸盐载体为0.0001mmol至5mmol,优选
0.001mmol至0.5mmol,并且更优选0.01至0.1mmol。进一步,在使用有机铝化合物的情况下,期望al的负载量在0.01mol至100mol、优选0.1mol至50mol、更优选0.2mol至10mol的范围内。
[0211]
对于负载和溶剂除去的方法,可以使用与以上无机载体的情况中相同的条件。当使用层状硅酸盐作为用作组分(b)和组分(c)二者的组分而使用时,所得乙烯系聚合物具有窄的分子量分布。此外,聚合活性高并且改善了具有长支链的乙烯系聚合物的生产性。由此获得的烯烃聚合用催化剂根据需要可以在单体的预聚合进行之后使用。
[0212]
5.烯烃系聚合物的生产方法
[0213]
上述烯烃聚合用催化剂可以用于烯烃聚合,特别是乙烯的均聚或乙烯与α
‑
烯烃的共聚。
[0214]
在本发明的烯烃系聚合物的生产方法中,优选至少包含乙烯,并且优选的是聚合物实质上为包含乙烯均聚物和乙烯与α
‑
烯烃的共聚物的乙烯系聚合物。
[0215]
作为共聚单体的α
‑
烯烃类包括碳数为3至30、优选3至8的那些,并且具体地,可以示例丙烯、1
‑
丁烯、1
‑
己烯、1
‑
辛烯和4
‑
甲基
‑1‑
戊烯等。关于α
‑
烯烃类,也可以使两种以上的α
‑
烯烃与乙烯共聚。共聚可以是任意的交替共聚、无规共聚和嵌段共聚。在使乙烯与其它α
‑
烯烃共聚的情况下,其它α
‑
烯烃的量可以任意地选自全部单体的90mol%以下的范围,但是一般选自40mol%以下、优选30mol%以下、进一步优选10mol%以下的范围。当然,也可以使用少量的除了乙烯和α
‑
烯烃以外的共聚单体。在该情况下,可以提及具有聚合性双键的化合物,例如,苯乙烯类,如苯乙烯、4
‑
甲基苯乙烯和4
‑
二甲基氨基苯乙烯;二烯类,如1,4
‑
丁二烯、1,5
‑
己二烯、1,4
‑
己二烯和1,7
‑
辛二烯;环状化合物,如降冰片烯和环戊烯;和含氧化合物类,如己烯醇、己烯酸和辛烯酸甲酯等。
[0216]
在本发明中,可以在上述负载型催化剂的存在下优选通过浆液聚合或气相聚合进行聚合反应。在浆液聚合的情况下,在氧和水等实质上不存在的状态下,在选自例如异丁烯、己烷和庚烷等脂肪族烃,例如苯、甲苯和二甲苯等芳香族烃,例如环己烷和甲基环己烷等脂环族烃的惰性烃溶剂的存在或不存在下,使乙烯等聚合。不必说的是,例如液态乙烯或液态丙烯等液态单体也可以用作溶剂。此外,在气相聚合的情况下,使乙烯等在向其中导入乙烯和共聚单体的气流并且使其流通或循环的反应器内聚合。在本发明中,进一步优选的聚合为气相聚合。关于聚合条件,温度为0℃至250℃,优选20℃至110℃,并且更优选60℃至100℃,并且当温度为60℃至90℃时,存在引入大量的长支链的倾向。此外,压力在常压至10mpa、优选常压至4mpa、更优选0.5mpa至2mpa的范围内。作为聚合时间,通常采用5分钟至10小时,优选5分钟至5小时。
[0217]
可以通过改变例如聚合温度和催化剂的摩尔比等聚合条件在一定程度上调节生成的聚合物的分子量,但是分子量的调节可以通过向聚合反应体系中添加氢气而更有效地进行。
[0218]
此外,即使当将用于水分除去目的的组分(所谓的清除剂)添加至聚合体系中时,仍可以进行聚合而没有任何困难。作为这样的清除剂,使用有机铝化合物,如三甲基铝、三乙基铝和三异丁基铝;上述有机铝氧化合物;包含支链烷基的改性的有机铝化合物;有机锌化合物,如二乙基锌和二丁基锌;有机镁化合物,如二乙基镁、二丁基镁和乙基丁基镁;格利雅化合物(grignard compounds),如乙基氯化镁和丁基氯化镁。其中,优选的是三乙基铝、
三异丁基铝和乙基丁基镁,并且特别优选的是三乙基铝。其也可以应用于具有其中例如氢气浓度、单体的量、聚合压力和聚合温度等聚合条件彼此不同的两个以上的阶段的多阶段聚合方式而没有任何困难。
[0219]
6.乙烯系聚合物的物理性质
[0220]
使用本发明的烯烃聚合用催化剂生产的烯烃系聚合物、特别是乙烯系聚合物的特征在于,引入了充分数量和长度的长支链并且更加改善了成形加工性。
[0221]
一般地,通过例如膜成形、吹塑成形或发泡成形等经由熔融状态的成形方法将聚乙烯加工成工业产品。在这种情况下,公知的是伸长流动性(elongation flowing property)大大影响成形的容易性。即,具有窄的分子量分布并且不具有长支链的聚乙烯具有低的熔融强度并且因此显示差的成形性。另一方面,具有超高分子量组分或长支链组分的聚乙烯的成形加工性优异。
[0222]
由通过将装配有差示折光计(ri)和粘度检测器(viscometer)的gpc设备与光散射检测器组合而测量的分子量为100,000和1,000,000时的支化指数(g
′
)的值认识到的是,由本发明的烯烃用聚合催化剂生产的乙烯系聚合物具有引入其中的充分数量和长度的长支链,并且在成形加工性方面优异。
[0223]
另外,在本说明书中,分子量为100,000和1,000,000时的支化指数(g
′
)的值分别称为“g
a
′”
和“g
b
′”
。
[0224]
此外,从成形加工性和机械特性优异的观点,本发明的乙烯系聚合物优选进一步具有以下特性。
[0225]
(1)mfr
[0226]
本发明中的乙烯系聚合物的mfr(熔体流动速率,190℃,2.16kg的负荷)优选为0.001g/10分钟至1,000g/10分钟,更优选0.01g/10分钟至100g/10分钟,进一步优选0.05g/10分钟至50g/10分钟,并且特别优选0.1g/10分钟至50g/10分钟。
[0227]
另外,乙烯系聚合物的mfr为根据jis k6760(190℃,2.16kg的负荷)测量的值。
[0228]
(2)密度
[0229]
本发明中的乙烯系聚合物的密度优选为0.85g/cm3至0.97g/cm3,更优选0.88g/cm3至0.95g/cm3,并且进一步优选0.90g/cm3至0.94g/cm3。
[0230]
另外,乙烯系聚合物的密度为根据jis k7112测量的值。
[0231]
(3)mw/mn
[0232]
本发明中的乙烯系聚合物的分子量分布(mw/mn)优选为2.0至10.0,更优选2.0至9.0,进一步优选2.5至8.0,并且特别优选2.5至7.5。
[0233]
另外,乙烯系聚合物的分子量分布(mw/mn)由重均分子量(mw)与数均分子量(mn)的比(mw/mn)来定义,并且为通过凝胶渗透色谱(gpc)法在以下条件下测量的值。
[0234]
从保留容积向分子量的换算使用由标准聚苯乙烯预先制成的校正曲线来进行。使用的标准聚苯乙烯均由tosoh corporation在以下品牌名称下生产:
[0235]
f380、f288、f128、f80、f40、f20、f10、f4、f1、a5000、a2500、a1000。通过注入0.2ml的通过将各标准聚苯乙烯溶解在odcb(包含0.5mg/ml的bht)中以获得0.5mg/ml的浓度而制备的溶液来创建校正曲线。作为校正曲线,使用根据最小二乘法通过近似获得的三次式。在用于向分子量的换算的粘度式[η]=k
×
m
α
中,使用以下数值。
[0236]
ps:k=1.38
×
10
‑4,α=0.7
[0237]
pe:k=3.92
×
10
‑4,α=0.733
[0238]
pp:k=1.03
×
10
‑4,α=0.78
[0239]
另外,gpc的测量条件如下。
[0240]
设备:由waters co.制造的gpc(alc/gpc 150c)
[0241]
检测器:由foxboro co.制造的miran 1a ir检测器(测量波长:3.42μm)
[0242]
柱:由showa denko k.k.制造的ad806m/s(3个柱)
[0243]
移动相溶剂:邻二氯苯
[0244]
测量温度:140℃
[0245]
流速:1.0ml/分钟
[0246]
注入量:0.2ml
[0247]
样品的制备:关于样品,使用odcb(包含0.5mg/ml的bht)来制备1mg/ml的溶液,并且在140℃下花费约1小时的时间来实现溶解。另外,如图1中所示例,取所得色谱图的基线和区间。
[0248]
(4)支化指数(g
a
′
和g
b
′
)
[0249]
在本发明的乙烯系聚合物中,分子量为100,000时的支化指数(g
a
′
)优选为0.50至0.99,更优选0.50至0.94,进一步优选0.50至0.87,并且还进一步优选0.55至0.80。在支化指数(g
a
′
)落入以上范围内的情况下,获得了在伸长粘度行为与熔融流动性之间具有优异的平衡的乙烯系聚合物。
[0250]
在本发明的乙烯系聚合物中,g
b
′
为0.30至0.75,优选0.30至0.68,更优选0.35至0.55,并且进一步优选0.35至0.50。当g
b
′
值大于0.75时,存在由于乙烯系聚合物的成形加工性不充分及其透明性不足而导致的不优选的情况。当g
b
′
值小于0.30时,乙烯系聚合物的成形加工性提高但是成形体的冲击强度降低或透明性劣化,以致存在不优选的情况。
[0251]
另外,支化指数(g
a
′
和g
b
′
)为通过以下方法测量的值。
[0252]
(i)借助gpc
‑
vis的支链结构分析
[0253]
作为装配有差示折光计(ri)和粘度检测器(viscometer)的gpc设备,使用waters co.的alliance gpcv2000。此外,作为光散射检测器,使用wyatt technology co.的多角度激光光散射检测器(malls)dawn
‑
e。将检测器按照malls、ri和viscometer的顺序连接。移动相溶剂为1,2,4
‑
三氯苯(以0.5mg/ml的浓度添加抗氧化剂irganox1076)。流速为1ml/分钟。作为柱,连接并且使用两根tosoh corporation的gmhhr
‑
h(s)ht的柱。在样品注入口处和在各检测器处的温度为140℃。样品浓度为1mg/ml。注入量(样品环容积)为0.2175ml。在求得从malls获得的绝对分子量(m)和惯性平方半径(inertial square radius)(rg)与从viscometer获得的特性粘度([η])时,利用连接至malls的数据处理软件astra(4.73.04版本)参考以下文献来进行计算。
[0254]
参考文献:
[0255]
1.聚合物特性的发展(developments in polymer characterization),第4卷,essex:应用科学(applied science);1984年,第1章。
[0256]
2.polymer,45,6495
‑
6505(2004)
[0257]
3.macromolecules,33,2424
‑
2436(2000)
[0258]
4.macromolecules,33,6945
‑
6952(2000)
[0259]
(ii)支化指数(g
a
′
和g
b
′
)的计算
[0260]
支化指数(g
a
′
)计算为通过借助以上viscometer测量样品获得的特性粘度(η支化)与通过单独测量线性聚合物获得的特性粘度(η线性)的比(η支化/η线性)。
[0261]
当将长支链引入至聚合物分子中时,与具有相同分子量的线性聚合物分子相比,惯性半径减小。由于在惯性半径减小时特性粘度降低,因此支化聚合物的特性粘度(η支化)与具有相同分子量的线性聚合物的特性粘度(η线性)的比(η支化/η线性)随着引入长支链而降低。因此,其中支化指数(g
a
′
=η支化/η线性)变为小于1的值的情况意味着引入了支链,并且其意味着引入的长支链随着所述值减小而增加。
[0262]
图2示出了借助以上gpc
‑
vis的分析结果的一个实例。图2表示分子量(m)时的支化指数(g
a
′
)。将logm=5时的g
′
值取作g
a
′
并且将logm=6时的g
′
值取作g
b
′
。这里,作为线性聚合物,使用直链聚乙烯标准参考物质1475a(standard reference material 1475a)(美国国家标准与技术研究院(national institute of standards&technology))。
[0263]
实施例
[0264]
以下参考实施例具体地描述本发明,但是本发明不应当解释为限于这些实施例。另外,用于实施例的评价方法如下。所有以下催化剂合成步骤和聚合步骤均在纯化的氮气气氛下进行,并且将使用的溶剂用分子筛4a脱水并且纯化。
[0265]
1.各种评价(测量)方法
[0266]
(1)mfr:
[0267]
根据jis k6760在190℃、2.16kg的负荷下测量。由作为在190℃、10kg的负荷的条件下同样测量的mfr的mfr10kg与mfr的比(=mfr10kg/mfr)来计算fr(流量比)。
[0268]
(2)分子量分布(mw/mn)的测量:
[0269]
通过在前述“6.乙烯系聚合物的物理性质”的“(3)mw/mn”项中记载的方法测量。
[0270]
(3)支化指数(g
′
)的测量:
[0271]
通过在前述“6.乙烯系聚合物的物理性质”的“(4)支化指数(g
a
′
和g
b
′
)”项中记载的方法测量。
[0272]
2.使用材料
[0273]
[茂金属化合物的合成]
[0274]
(1)茂金属化合物a:二甲基亚甲硅基(2,5
‑
二甲基
‑3‑
苯基
‑
环戊二烯并(cyclopento)[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0275]
(1
‑
1)2,5
‑
二甲基
‑3‑
苯基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成
[0276]
2,5
‑
二甲基
‑3‑
苯基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成根据在j.am.chem.soc.2001,123,4763
‑
4773的实验项中记载的过程来进行。
[0277]
(1
‑
2)(2,5
‑
二甲基
‑3‑
苯基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的合成
[0278]
在将3.48g(15.38mmol)的2,5
‑
二甲基
‑3‑
苯基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和35ml的thf添加至100ml的烧瓶中以形成溶液之后,将其冷却至
‑
78℃并且将7.38ml(18.5mmol)的正丁基锂/己烷溶液(2.5m)添加至其中,接着在10℃下搅拌3小时。将3.97g(30.76mmol)的二甲基二氯硅烷和20ml的thf添加至另外准备的100ml的烧瓶中,并且将整
体冷却至
‑
78℃并将先前的反应溶液添加至其中。将整体在10℃下搅拌12小时。通过借助减压蒸馏除去挥发物而获得黄色溶液。将58.5ml的thf添加至黄色溶液中,由此形成溶液,并且在
‑
30℃下将8.08ml(16.2mmol)的环戊二烯基钠/thf溶液(2m)缓慢地添加至其中。将整体在10℃下搅拌90分钟。将反应产物缓慢的添加至100ml的冰水中,用100ml的乙酸乙酯萃取两次,将所得有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得2.97g(收率55%)的(2,5
‑
二甲基
‑3‑
苯基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的黄色油。
[0279]
(1
‑
3)二甲基亚甲硅基(2,5
‑
二甲基
‑3‑
苯基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0280]
将2.97g(8.52mmol)的(2,5
‑
二甲基
‑3‑
苯基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷和30ml的二乙醚添加至200ml的烧瓶中,接着冷却至
‑
78℃。将7.16ml(17.9mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,并且使温度恢复至室温,接着搅拌3小时。将反应溶液的溶剂通过减压蒸馏除去,将90ml的二氯甲烷添加至其中,并且将整体冷却至
‑
78℃。将2.08g(8.95mmol)的四氯化锆添加至其中,并且在使温度缓慢地恢复至室温的同时将整体搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从滤液中除去溶剂,由此获得黄色粉末。将粉末用18ml的甲苯和6ml的正戊烷的混合溶剂洗涤,从而获得0.98g(收率22%)的甲基亚甲硅基(2,5
‑
二甲基
‑3‑
苯基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的黄色晶体。
[0281]1h
‑
nmr值(cdcl3):δ0.90(s,3h),δ0.95(s,3h),δ2.24(s,3h),δ2.55(s,3h),δ5.94(t,2h),δ6.55(s,1h),δ6.92(m,2h),δ7.34(m,1h),δ7.45(t,2h),δ7.53(m,2h)。
[0282]
(2)茂金属化合物b:二甲基亚甲硅基(3
‑
苯基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0283]
(2
‑
1)4
‑
苯基噻吩
‑2‑
羧基醛(carboxyaldehyde)的合成
[0284]
在氮气下将2.00g(11.49mmol)的4
‑
溴噻吩
‑2‑
羧基醛、1.40g(11.49mmol)的苯基硼酸、2.44g(22.98mmol)的碳酸钠、15ml的甲苯、15ml的乙醇和5ml的水添加至100ml的烧瓶中之后,将663.96mg的pd(pph3)4添加至其中并且将整体在100℃下搅拌6小时。用100ml的乙酸乙酯进行萃取两次,并且将所得有机相用50ml的水和50ml的饱和食盐水洗涤,并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.60g(收率81.3%)的4
‑
苯基噻吩
‑2‑
羧基醛的黄色固体。
[0285]
(2
‑
2)3
‑
(4
‑
苯基
‑2‑
噻吩基)
‑
丙酸的合成
[0286]
在氮气下,将29.53g(256.25mmol)的三乙胺添加至300ml的烧瓶中并且将30.78g(640.64mmol)的甲酸在0℃下缓慢地添加至其中。在将混合物在25℃下搅拌1.5小时之后,将15ml的dmf添加至其中。随后,添加16.16g(112.11mmol)的2,2
‑
二甲基
‑
1,3
‑
二噁烷
‑
4,6
‑
二酮的dmf(100ml)溶液,并且进一步,将20.10g(106.77mmol)的4
‑
苯基噻吩
‑2‑
羧基醛在25℃下添加至其中。当将混合物在100℃下搅拌72小时时,其变为棕色溶液。将反应溶液添加至150ml的冰水中并且用15ml的6n盐酸将ph调节至1,由此使固体析出。将析出的固体通过过滤收集并且用20ml的水洗涤五次并用100ml的乙酸乙酯洗涤两次,从而获得22.67g(收率91.40%)的3
‑
(4
‑
苯基
‑2‑
噻吩基)
‑
丙酸的黄色固体。
[0287]
(2
‑
3)3
‑
苯基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮的合成
[0288]
将20.60g(88.68mmol)的3
‑
(4
‑
苯基
‑2‑
噻吩基)
‑
丙酸添加至100ml的烧瓶中,并且在78至83℃下将23.06g(15.17ml,96.86mmol)的eaton试剂添加至其中。在将混合物在80℃下搅拌1小时之后,将其倒入200ml的冰水中并且用200ml的二氯甲烷进行萃取三次。将所得有机相用200ml的碳酸钠水溶液洗涤并且经无水硫酸钠干燥。过滤硫酸钠,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,由此获得8.20g(收率43.2%)的3
‑
苯基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮的淡黄色固体。
[0289]
(2
‑
4)3
‑
苯基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成
[0290]
在氮气下,将3.00g(14.00mmol)的3
‑
苯基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮和50ml的无水thf添加至100ml的烧瓶中,并且在0℃下将425.04mg(11.20mmol)的氢化铝锂添加至其中。在将混合物在20℃下搅拌1.5小时之后,将反应产物倒入20ml的冰水中并且用50ml的乙酸乙酯萃取三次。将所得有机相用50ml的饱和氯化铵水溶液洗涤两次并且经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得2.85g的黄色固体。
[0291]
在氮气下,将2.85g的以上黄色固体和150ml的甲苯添加至300ml的烧瓶中,并且将25.06mg(131.80μmol)的对甲苯磺酸一水合物和27.19mg(131.80μmol)的2,6
‑
二
‑
叔丁基苯酚添加至其中。当将混合物在110℃下搅拌2小时时,获得淡黄色溶液。将所得淡黄色溶液倒入50ml的饱和碳酸氢钠水溶液中并且将有机相分离。将水相用30ml的乙酸乙酯萃取三次,将所得有机相与先前的有机相混合,并且将混合的有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.15g(收率44.0%)的3
‑
苯基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的淡黄色油。
[0292]
(2
‑
5)(3
‑
苯基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的合成
[0293]
在氮气下将1.10g(5.55mmol)的3
‑
苯基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和30ml的thf添加至100ml的烧瓶中以形成溶液之后,将其冷却至
‑
78℃并且添加2.66ml(6.65mmol)的正丁基锂/己烷溶液(2.5m),接着在10℃下搅拌3小时。将1.43g(11.10mmol)的二甲基二氯硅烷和10ml的thf添加至另外准备的100ml的烧瓶中,并且将整体冷却至
‑
78℃并将先前的反应溶液添加至其中。在将整体在10℃下搅拌12小时之后,通过借助减压蒸馏除去挥发物而获得1.61g的淡黄色固体。将30ml的thf添加至该淡黄色固体中,由此形成溶液,并且在
‑
30℃下将3.05ml(6.1mmol)的环戊二烯基钠/thf溶液(2m)添加至其中。将整体在10℃下搅拌90分钟。将反应产物添加至20ml的冰水中并且将有机相分离。将水相用50ml的乙酸乙酯萃取两次,将所得有机相与先前的有机相混合,并且将混合的有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得690mg(收率38.8%)的(3
‑
苯基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的淡黄色油。
[0294]
(2
‑
6)二甲基亚甲硅基(3
‑
苯基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0295]
将2.50g(7.80mmol)的(3
‑
苯基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷和50ml的二乙醚添加至200ml的烧瓶中,接着冷却至
‑
78℃。将6.55ml(16.4mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,接着在10℃下搅拌3小时。将反应溶液的溶剂通过减压蒸馏而除去,将90ml的二氯甲烷添加至其中,并且将整体冷却至
‑
78
℃。将1.91g(8.18mmol)的四氯化锆添加至其中,使温度经3小时升高至10℃,并且将整体在10℃下进一步搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从所得滤液中除去溶剂,由此获得黄色粉末。将粉末用11ml的甲苯和4ml的正戊烷的混合溶剂洗涤并且进一步用90ml的二氯甲烷萃取从而除去不溶物。将所得二氯甲烷溶液通过减压蒸馏进行除去,由此获得0.9g(收率24%)的二甲基亚甲硅基(3
‑
苯基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的黄色晶体。
[0296]1h
‑
nmr值(cdcl3):δ0.86(s,3h),δ0.91(s,3h),δ5.94(m,1h),δ6.00(m,1h),δ6.14(m,1h),δ6.90(m,1h),δ6.95(m,1h),δ7.04(d,1h),δ7.35(t,1h),δ7.44(t,2h),δ7.52(d,1h),δ7.66(d,2h)。
[0297]
(3)茂金属化合物c:二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0298]
(3
‑
1)4
‑
溴噻吩
‑2‑
羧基醛的合成
[0299]
在氮气下,将17.00g(70.27mmol)的3,4
‑
二溴噻吩和35ml的二乙醚添加至300ml的烧瓶中,并且在
‑
78℃下缓慢地滴加28.11ml(70.28mmol)的正丁基锂/正己烷溶液(2.5m),接着仍在
‑
78℃下搅拌15分钟。随后,将5.14g(70.27mmol)的dmf添加至其中并且将整体在
‑
78℃下搅拌3小时。将反应溶液升温至15℃并且将40ml的水添加至其中。用50ml的乙酸乙酯进行萃取两次并且经碳酸钾干燥有机相。将碳酸钾过滤并且将溶液通过减压蒸馏进行除去,从而获得15g(纯度约80%)的4
‑
溴噻吩
‑2‑
羧基醛的粗产物。
[0300]
(3
‑
2)3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丙酸的合成
[0301]
在氮气下,将7.54g(157.02mmol)的甲酸添加至300ml的烧瓶中,并且将6.36g(62.81mmol)的三乙胺在0℃下缓慢地添加至其中。在将混合物在20℃下搅拌1.5小时之后,将50ml的dmf添加至其中。随后,添加3.96g(27.48mmol)的2,2
‑
二甲基
‑
1,3
‑
二噁烷
‑
4,6
‑
二酮的dmf(10ml)溶液,并且进一步,将5.00g(26.17mmol)的4
‑
溴噻吩
‑2‑
羧基醛的dmf(10ml)溶液在20℃下添加至其中。当将混合物在100℃下搅拌12小时时,其变为棕色溶液。将反应溶液添加至100ml的冰水中并且用2n氢氧化钠水溶液将ph调节至12。用100ml的二氯甲烷进行萃取两次,并且将水相用6n盐酸调节至ph 1并且再次用100ml的二氯甲烷萃取三次。将有机相经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得4.30g的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丙酸的粗产物。
[0302]
(3
‑
3)3
‑
溴
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的合成
[0303]
将111.38g(73.28ml,467.89mmol)的eaton试剂添加至200ml的烧瓶中,并且在40℃下将11.00g(46.79mmol)的3
‑
(4
‑
溴
‑2‑
噻吩基)
‑
丙酸添加至其中。在将混合物在40℃下搅拌30分钟之后,将其倒入200ml的冰水中并且用200ml的二氯甲烷进行萃取四次。将所得有机相用200ml的饱和碳酸氢钠水溶液洗涤并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.90g(收率18.7%)的3
‑
溴
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的黄色固体。
[0304]
(3
‑
4)3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的合成
[0305]
在氮气下将8.00g(36.85mmol)的3
‑
溴
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮、423.81mg(737.00μmol)的pd(dba)2、773.29mg(2.95mmol)的三苯基膦和200ml的甲苯添加至500ml的烧瓶之后,将另外准备的16.41g三(正丁基)(5
‑
甲基
‑2‑
呋喃基)锡的甲苯(40ml)溶液添加
至其中,接着在110℃下搅拌16小时。将反应溶液用300ml的乙酸乙酯稀释并且将稀释的溶液用200ml的5%氟化钾水溶液洗涤。在将有机相分离之后,将水相用200ml的乙酸乙酯萃取两次。将所得有机相与先前的有机相混合,将混合的有机相用300ml的饱和食盐水洗涤,并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,通过硅胶柱进行纯化,并且进一步从乙酸乙酯/石油醚的混合溶剂中进行重结晶,从而获得7.04g(收率87.5%)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的黄色固体。
[0306]
(3
‑
5)3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成
[0307]
在氮气下,将4.00g(18.33mmol)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮和40ml的乙醇添加至100ml的烧瓶中,并且在20℃下将3.58g(19.25mmol)的4
‑
甲基苯磺酰肼和697.19mg(3.67mmol)的甲苯磺酸一水合物添加至其中。将混合物在78℃下搅拌16小时,从而获得黄色的悬浮液。在冷却至0℃之后,将黄色固体通过过滤来回收并且用冷却至0℃的10ml的乙醇洗涤三次,然后将黄色固体在减压下干燥。
[0308]
在氮气下,将获得的黄色固体和300ml的thf添加至500ml的烧瓶中,并且在
‑
78℃下滴加16.04ml(40.1mmol)的正丁基锂/正己烷溶液(2.5m),接着在66℃下搅拌20分钟。将反应溶液倒入200ml的冰水中,并且在将有机相分离之后,将水相用200ml的乙酸乙酯萃取两次。将所得有机相与先前的有机相混合并且将混合的有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得2.27g(收率61.4%)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的黄色油。
[0309]
(3
‑
6)(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的合成
[0310]
在氮气下,将1.00g(4.94mmol)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和20ml的thf添加至100ml的烧瓶中,将整体冷却至
‑
78℃,并且添加2.37ml(5.93mmol)的正丁基锂/己烷溶液(2.5m),接着在20℃下搅拌3小时。随后,在
‑
78℃下将11.06ml(7.41mmol)的环戊二烯基二甲基甲硅烷基氯/己烷溶液(0.67m)缓慢地添加至其中,并且将整体在20℃下搅拌1.5小时。将反应溶液倒入150ml的冰水中并且用100ml的乙酸乙酯萃取四次。将有机相用饱和食盐水洗涤并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.04g(收率64.9%)的(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的淡黄色油。
[0311]
(3
‑
7)二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0312]
在氮气下,将1.20g(3.70mmol)的(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷和18ml的二乙醚添加至200ml的烧瓶中,接着冷却至
‑
78℃。将3.11ml(7.78mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,接着在20℃下搅拌3小时。将反应溶液的溶剂通过减压蒸馏除去,将50ml的二氯甲烷添加至其中,并且将整体冷却至
‑
78℃。将947.93mg(4.07mmol)的四氯化锆添加至其中,使温度经3小时升高至20℃,并且将整体在20℃下进一步搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从所得滤液除去溶剂,由此获得黄色粉末。将粉末用10ml的甲苯和5ml的正戊烷的混合溶剂洗涤并且进一步用50ml的甲苯萃取从而除去不溶物。将所得甲苯溶液通过减压蒸馏进行除去,由此获得270mg(收率15.1%)的二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的绿色固体。
[0313]1h
‑
nmr值(cdcl3):δ0.85(s,3h),δ0.89(s,3h),δ2.36(s,3h),δ5.92(d,1h),δ5.99(d,1h),δ6.07(d,1h),δ6.13(d,1h),δ6.50(d,1h),δ6.87(d,1h),δ6.93(d,1h),δ7.00(d,1h),δ7.56(s,1h)。
[0314]
(4)茂金属化合物d:二甲基亚甲硅基(3
‑
苯基
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0315]
(4
‑
1)3
‑
苯基
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成
[0316]
在氮气下,将在第(2)项茂金属化合物(b)的合成的(2
‑
3)中获得的8.00g(37.33mmol)的3
‑
苯基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮和100ml的甲苯添加至200ml的烧瓶中,并且在0℃下将18.67ml(56.01mmol)的甲基溴化镁(3m)添加至其中,接着在15℃下搅拌12小时。将反应溶液倒入100ml的冰水中,将有机相分离,并且经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得8.3g的棕色油。
[0317]
在氮气下,将5.00g的以上棕色油和500ml的甲苯添加至1000ml的烧瓶中,并且将41.29mg(217.1μmol)的甲苯磺酸一水合物和44.79mg(217.1μmol)的2,6
‑
二
‑
叔丁基苯酚添加至其中,接着在110℃下搅拌1小时。在冷却至室温之后,将反应溶液倒入100ml的饱和碳酸氢钠水溶液中。将有机相分离,将水相用100ml的乙酸乙酯萃取三次,将所得有机相与先前的有机相混合,并且将混合的有机相经无水硫酸钠干燥。通过硅胶柱进行纯化,从而获得2.27g(收率61.4%)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的黄色油。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.80g(收率39.1%)的3
‑
苯基
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的黄色油。
[0318]
(4
‑
2)(3
‑
苯基
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的合成
[0319]
在氮气下将3.70g(17.43mmol)的3
‑
苯基
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和50ml的thf添加至100ml的烧瓶中以形成溶液之后,将其冷却至
‑
78℃并且添加8.37ml(20.9mmol)的正丁基锂/己烷溶液(2.5m),接着在10℃下搅拌3小时。将整体再次冷却至
‑
78℃并且迅速地添加4.50g(34.86mmol)的二甲基二氯硅烷,接着在10℃下搅拌12小时。通过借助减压蒸馏除去挥发物,获得5.31g的淡黄色油。将60ml的thf添加至该淡黄色油中,由此形成溶液,并且在
‑
78℃下将9.14ml(18.3mmol)的环戊二烯基钠/thf溶液(2m)添加至其中。将整体在10℃下搅拌2小时,将反应产物添加至50ml的冰水中,并且将有机相分离。将水相用100ml的乙酸乙酯萃取两次,将所得有机相与先前的有机相混合,并且将混合的有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得2.20g(收率32.1%)的(3
‑
苯基
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的淡黄色油。
[0320]
(4
‑
3)二甲基亚甲硅基(3
‑
苯基
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0321]
在氮气下将2.20g(6.58mmol)的(3
‑
苯基
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷和50ml的二乙醚添加至200ml的烧瓶中,接着冷却至
‑
78℃。将5.53ml(13.8mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,接着在10℃下搅拌3小时。将反应溶液的溶剂通过减压蒸馏除去,将70ml的二氯甲烷添加至其中,并且将整体冷却
至
‑
78℃。将1.69g(7.24mmol)的四氯化锆添加至其中,使温度经3小时升高至10℃,并且将整体在10℃下进一步搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从所得滤液除去溶剂,由此获得黄色固体。将固体用11ml的甲苯和4ml的正戊烷的混合溶剂洗涤,然后在减压下干燥,由此获得1.40g(收率43.0%)的二甲基亚甲硅基(3
‑
苯基
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的淡绿色固体。
[0322]1h
‑
nmr值(cdcl3):δ0.80(s,3h),δ0.90(s,3h),δ2.17(s,3h),δ5.62(s,1h),δ5.81(q,1h),δ5.94(q,1h),δ6.95(m,1h),δ7.01(m,1h),δ7.28(s,1h),δ7.36
‑
7.40(m,3h),δ7.49(d,2h)。
[0323]
(5)茂金属化合物e:二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0324]
(5
‑
1)3
‑
乙酰基
‑4‑
溴噻吩的合成
[0325]
在氮气下,将40.00g(165.34mmol)的3,4
‑
二溴噻吩和200ml的二乙醚添加至500ml的烧瓶中,并且在
‑
78℃下将66.14ml(165.4mmol)的正丁基锂/正己烷溶液(2.5m)缓慢地滴加至其中,接着仍在
‑
78℃下搅拌30分钟。随后,在
‑
78℃下将20.46g(198.41mmol)的n
‑
甲氧基
‑
n
‑
甲基
‑
乙酰胺添加至其中并且将整体在20℃下搅拌12小时。将反应溶液用2n盐酸调节至ph=7并且用100ml的乙酸乙酯萃取三次。将有机相用100ml的水洗涤并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得19.0g(收率56.0%)的3
‑
乙酰基
‑4‑
溴噻吩的淡黄色油。
[0326]
(5
‑
2)3
‑
(4
‑
溴
‑3‑
噻吩基)
‑3‑
羟基丁酸乙酯的合成
[0327]
在氮气下,将22.96g(351.10mmol)的锌和180ml的thf添加至500ml的烧瓶中,并且在20℃下将3.18g(29.26mmol)的三甲基甲硅烷基氯添加至其中,接着搅拌30分钟。将混合物加热至40℃,将29.32g(175.55mmol)的溴乙酸乙酯缓慢地添加至其中,并且将整体在40℃下搅拌2小时,从而获得棕色的悬浮液。在氮气下,将30.00g(146.29mmol)的3
‑
乙酰基
‑4‑
溴噻吩和80ml的thf添加至另外准备的500ml的烧瓶中,并且在66℃下将先前的棕色的悬浮液缓慢地添加至其中,接着仍在66℃下搅拌6小时。将反应溶液倒入400ml的冰水中并且用200ml的乙酸乙酯萃取两次。将有机相用100ml的水洗涤三次并且经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得40.00g的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑3‑
羟基丁酸乙酯的棕色油。
[0328]
(5
‑
3)3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丁酸乙酯的合成
[0329]
在氮气下,将40.00g(136.43mmol)的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑3‑
羟基丁酸乙酯、19.04g(163.72mmol)的三乙基硅烷和308.02g(2.70mol)的三氟乙酸添加至1000ml的烧瓶中,接着在71℃下搅拌4小时。将所得溶液浓缩并且将浓缩物溶解在500ml的乙酸乙酯中,并且将反应溶液用碳酸钠水溶液调节至ph=7。将有机层分离,用100ml的水洗涤三次,并且经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得55.00g的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丁酸乙酯的棕色油。
[0330]
(5
‑
4)3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丁酸的合成
[0331]
在氮气下,将50.00g(180.39mmol)的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丁酸乙酯和300ml的乙醇添加至1000ml的烧瓶中,并且在20℃下将40.49g(721.56mmol)的氢氧化钾添加至其中,接着在78℃下搅拌12小时。将反应溶液浓缩,添加300ml的水,并且用100ml的乙酸乙酯进行
萃取三次。将水相用6n盐酸调节至ph=3并且用200ml的二氯甲烷萃取三次。将有机相经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得25.00g的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丁酸的棕色油。
[0332]
(5
‑
5)3
‑
溴
‑4‑
甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的合成
[0333]
在氮气下,将30.00g(120.42mmol)的3
‑
(4
‑
溴
‑3‑
噻吩基)
‑
丁酸添加至500ml的烧瓶中,并且在20℃下将246.00g(2.07mol)的亚硫酰氯添加至其中。将混合物在76℃下搅拌3小时并且通过减压蒸馏除去未反应的亚硫酰氯。随后,加入200ml的二氯甲烷并且在0℃下添加19.14g(143.51mmol)的氯化铝,接着在20℃下搅拌2小时。将反应溶液倒入200ml的冰水中并且用200ml的二氯甲烷进行萃取三次。将有机相用100ml的水洗涤三次并且经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得24.50g的3
‑
溴
‑4‑
甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的棕色油。
[0334]
(5
‑
6)3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的合成
[0335]
在氮气下将13.50g(58.41mmol)的3
‑
溴
‑4‑
甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮、671.77mg(1.17mmol)的pd(dba)2、1.23g(4.67mmol)的三苯基膦和250ml的甲苯添加至500ml的烧瓶之后,将另外准备的26.02g(70.10mmol)的三(正丁基)(5
‑
甲基
‑2‑
呋喃基)锡添加至其中,接着在110℃下搅拌12小时。将反应溶液倒入400ml的5%氟化钾水溶液中并且用200ml的乙酸乙酯进行萃取三次。在用100ml的水洗涤三次之后,将有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得9.30g(收率68.54%)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮的淡黄色油。
[0336]
(5
‑
7)3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成
[0337]
在氮气下,将5.00g(21.52mmol)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑6‑
酮和40ml的乙醇添加至100ml的烧瓶中,并且在20℃下将4.81g(25.82mmol)的4
‑
甲基苯磺酰肼和1.02g(5.38mmol)的甲苯磺酸一水合物添加至其中。将混合物在78℃下搅拌18小时,从而获得黄色的悬浮液。将黄色固体通过过滤来回收,用10ml的乙醇洗涤两次,然后在减压下干燥,从而获得4.6g的黄色固体。将该反应再进行一次,从而总共获得6.8g的黄色固体。
[0338]
在氮气下,将5.00g的获得的黄色固体和60ml的thf添加至200ml的烧瓶中,并且在
‑
78℃下滴加12.48ml(31.20mmol)的正丁基锂/正己烷溶液(2.5m),接着在66℃下搅拌1小时。将反应溶液倒入80ml的冰水中,用60ml的乙酸乙酯进行萃取三次,并且将所得有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.60g(收率59.3%)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的黄色油。
[0339]
(5
‑
8)(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的合成
[0340]
在氮气下,将2.00g(9.25mmol)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和20ml的thf添加至100ml的烧瓶中,将整体冷却至
‑
78℃,并且添加4.07ml(10.18mmol)的正丁基锂/己烷溶液(2.5m),接着在20℃下搅拌3小时。随后,在
‑
78℃下将15.19ml(10.18mmol)的环戊二烯基二甲基甲硅烷基氯/己烷溶液(0.67m)缓慢地添加至其
中,并且将整体在20℃下搅拌1.5小时。将反应溶液倒入150ml的冰水中并且用100ml的乙酸乙酯萃取四次。将有机相用饱和食盐水洗涤并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.46g(收率46.6%)的(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷的黄色油。
[0341]
(5
‑
9)二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0342]
在氮气下,将1.46g(4.31mmol)的(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二甲基硅烷和20ml的二乙醚添加至200ml的烧瓶中,接着冷却至
‑
78℃。将3.62ml(9.05mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,接着在20℃下搅拌3小时。将反应溶液的溶剂通过减压蒸馏除去,将55ml的二氯甲烷添加至其中,并且将整体冷却至
‑
78℃。将1.10g(4.74mmol)的四氯化锆添加至其中,使温度经3小时升高至20℃,并且将整体在20℃下进一步搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从所得滤液除去溶剂,由此获得棕色的粉末。将粉末用5ml的甲苯和10ml的正戊烷的混合溶剂洗涤并且进一步用100ml的甲苯萃取从而除去不溶物。将所得甲苯溶液通过减压蒸馏进行除去,由此获得207mg(收率9.67%)的二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的绿色固体。
[0343]1h
‑
nmr值(cdcl3):δ0.80(s,3h),δ0.89(s,3h),δ2.35(s,3h),δ2.49(s,3h),δ5.65(s,1h),δ5.81
‑
5.83(m,1h),δ5.91
‑
5.93(m,1h),δ6.03
‑
6.05(m,1h),δ6.45(d,1h),δ6.91
‑
6.94(m,1h),δ6.96
‑
6.99(m,1h),δ7.47(s,1h)。
[0344]
(6)茂金属化合物f:二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(2,3,4,5
‑
四甲基环戊二烯基)二氯化锆的合成
[0345]
(6
‑
1)(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(2,3,4,5
‑
四甲基环戊二烯基)二甲基硅烷的合成
[0346]
在氮气下,将在第(5)项茂金属化合物e的合成的(5
‑
7)中获得的2.50g(11.56mmol)的3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和40ml的thf添加至100ml的烧瓶中,将整体冷却至
‑
78℃,并且添加5.78ml(14.45mmol)的正丁基锂/己烷溶液(2.5m),接着在20℃下搅拌3小时。随后,在
‑
78℃下将22.94ml(15.37mmol)的环戊二烯基二甲基甲硅烷基氯/己烷溶液(0.67m)缓慢地添加至其中,并且将整体在20℃下搅拌1.5小时。将反应溶液倒入150ml的冰水中并且用150ml的乙酸乙酯萃取三次。将有机相用饱和食盐水洗涤并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得2.48g(收率54.4%)的(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(2,3,4,5
‑
四甲基环戊二烯基)二甲基硅烷的黄色油。
[0347]
(6
‑
2)二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(2,3,4,5
‑
四甲基环戊二烯基)二氯化锆的合成
[0348]
在氮气下,将1.06g(2.69mmol)的(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑6‑
氢环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(2,3,4,5
‑
四甲基环戊二烯基)二甲基硅烷和20ml的二乙醚添加至100ml的烧瓶中,接着冷却至
‑
78℃。将2.64ml(6.60mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,接着在20℃下搅拌3小时。将反应溶液的溶剂通过减压蒸馏除去,将40ml的二氯
甲烷添加至其中,并且将整体冷却至
‑
78℃。将689.57mg(2.96mmol)的四氯化锆添加至其中,使温度经3小时升高至20℃,并且将整体在20℃下进一步搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从所得滤液除去溶剂,由此获得黄色粉末。将粉末用15ml的甲苯和25ml的正戊烷的混合溶剂洗涤并且进一步用150ml的甲苯萃取以除去不溶物。将所得甲苯溶液通过减压蒸馏进行除去,由此获得409mg(收率27.4%)的二甲基亚甲硅基(3
‑
(5
‑
甲基
‑2‑
呋喃基)
‑4‑
甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(2,3,4,5
‑
四甲基环戊二烯基)二氯化锆的黄色固体。
[0349]1h
‑
nmr值(cdcl3):δ0.90(s,3h),δ1.02(s,3h),δ1.93(s,3h),δ2.00(s,3h),δ2.05(s,3h),δ2.08(s,3h),δ2.35(s,3h),δ2.48(s,3h),δ5.33(s,1h),δ6.02(s,1h),δ6.39(s,1h),δ7.44(s,1h)。
[0350]
(7)茂金属化合物g:二甲基亚甲硅基(2,5
‑
二甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑6‑
基)(环戊二烯基)二氯化锆的合成
[0351]
(7
‑
1)2,5
‑
二甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮的合成
[0352]
将152.04g(638.69mmol)的eaton试剂添加至300ml的烧瓶中,并且在78至83℃下将10.00g(101.86mmol)的2
‑
甲基噻吩和10.52g(122.24mmol)的甲基丙烯酸的混合物经30分钟添加至其中。在将混合物在80℃下搅拌5分钟之后,将其逐步倒入200ml的冰水中并且用300ml的二氯甲烷进行萃取。将有机相分离并且将水相用300ml的二氯甲烷萃取两次。将所得有机相合并并且将合并的有机相用300ml的饱和碳酸钠水溶液洗涤两次并且经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得4.90g(收率28%)的2,5
‑
二甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮的黄色液体。
[0353]
(7
‑
2)2,5
‑
二甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的合成
[0354]
在氮气下,将4.90g(29.48mmol)的2,5
‑
二甲基
‑
环戊烯并[2,3
‑
b]噻吩
‑4‑
酮和50ml的无水thf添加至100ml的烧瓶中,并且在0℃下将1.34g(35.38mmol)的氢化铝锂添加至其中。在将混合物在15℃下搅拌2小时之后,将反应产物缓慢地倒入100ml的冰水中并且用300ml的乙酸乙酯萃取两次。将所得有机相经无水硫酸钠干燥。将硫酸钠过滤并且将溶液通过减压蒸馏进行除去,从而获得4.44g的黄色固体。
[0355]
在氮气下,将780mg的以上黄色固体和15ml的甲苯添加至50ml的烧瓶中,并且将44.13mg(232.00μmol)的对甲苯磺酸一水合物和9.57mg(46.40μmol)的2,6
‑
二
‑
叔丁基苯酚添加至其中。在将混合物在110℃下搅拌10分钟之后,使温度恢复至室温并且将混合物分别用50ml的饱和碳酸钠水溶液和50ml的水洗涤。将所得有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得460.00mg(收率66%)的2,5
‑
二甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩的黄色油。
[0356]
(7
‑
3)(2,5
‑
二甲基
‑4‑
氢环戊二烯并[2,3
‑
b]噻吩
‑4‑
基)(环戊二烯基)二甲基硅烷的合成
[0357]
在氮气下将2.06g(13.71mmol)的2,5
‑
二甲基
‑6‑
氢环戊二烯并[1,2
‑
b]噻吩和30ml的thf添加至100ml的烧瓶中以形成溶液之后,将其冷却至
‑
78℃并且添加6.58ml(16.5mmol)的正丁基锂/己烷溶液(2.5m),接着在15℃下搅拌3小时。将3.54g(27.42mmol)的二甲基二氯硅烷和10ml的thf添加至另外准备的100ml的烧瓶中,并且将整体冷却至
‑
78℃并将先前的反应溶液添加至其中。在将整体在10℃下搅拌12小时之后,通过借助减压蒸
馏除去挥发物而获得3.33g的黄色油。将30ml的thf添加至2.57g的黄色油中,由此形成溶液,并且在
‑
78℃下将6.18ml(12.4mmol)的环戊二烯基钠/thf溶液(2m)添加至其中。将整体在15℃下搅拌2小时。将反应产物添加至50ml的冰水中并且将有机相分离。将水相用100ml的乙酸乙酯萃取两次,将所得有机相与先前的有机相混合,并且将混合的有机相经无水硫酸钠干燥。将硫酸钠过滤,将溶液通过减压蒸馏进行除去,并且通过硅胶柱进行纯化,从而获得1.76g(收率57.6%)的(2,5
‑
二甲基
‑4‑
氢环戊二烯并[2,3
‑
b]噻吩
‑4‑
基)(环戊二烯基)二甲基硅烷的黄色油。
[0358]
(7
‑
4)二甲基亚甲硅基(2,5
‑
二甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑4‑
基)(环戊二烯基)二氯化锆的合成
[0359]
在氮气下将2.40g(8.81mmol)的(2,5
‑
二甲基
‑4‑
氢环戊二烯并[2,3
‑
b]噻吩
‑4‑
基)(环戊二烯基)二甲基硅烷和30ml的二乙醚添加至200ml的烧瓶中,接着冷却至
‑
78℃。将7.40ml(18.5mmol)的正丁基锂/正己烷溶液(2.5m)滴加至其中,接着在15℃下搅拌3小时。
[0360]
将反应溶液的溶剂通过减压蒸馏除去,以获得2.50g的黄色固体。将80ml的二氯甲烷添加至1.95g(6.86mmol)的黄色固体中,并且将整体冷却至
‑
78℃。将1.68g(7.20mmol)的四氯化锆添加至其中,使温度经3小时升高至20℃,并且将整体在20℃下进一步搅拌过夜。将反应溶液过滤并且通过在减压下蒸馏从所得滤液除去溶剂,由此获得黄色粉末。将粉末用34ml的甲苯和11ml的正戊烷的混合溶剂萃取,从而除去不溶物。将所得溶液通过减压蒸馏进行除去,由此获得黄绿色固体。进一步,将固体用50ml的二氯甲烷萃取并且将所得溶液通过减压蒸馏进行除去,由此获得0.92g(收率31%)的二甲基亚甲硅基(2,5
‑
二甲基
‑
环戊二烯并[2,3
‑
b]噻吩
‑4‑
基)(环戊二烯基)二氯化锆的黄绿色固体。
[0361]1h
‑
nmr值(cdcl3):δ0.905(s,3h),δ0.913(s,3h),δ2.23(s,3h),δ2.45(s,3h),δ5.69(m,1h),δ5.97(m,1h),δ6.43(s,1h),δ6.55(s,1h),δ6.87(m,1h),δ6.95(m,1h)。
[0362]
(8)茂金属化合物h:二甲基亚甲硅基(4
‑
苯基
‑
茚基
‑1‑
基)(环戊二烯基)二氯化锆的合成
[0363]
关于二甲基亚甲硅基(4
‑
苯基
‑
茚基
‑1‑
基)(环戊二烯基)二氯化锆的合成,其根据jp
‑
a
‑
2011
‑
137146的实施例(合成例5)中记载的过程来合成。
[0364]
(实施例1)
[0365]
(1)固体催化剂的制备
[0366]
在氮气气氛下,将5g的在600℃下煅烧5小时的二氧化硅放入200ml的双颈烧瓶中,并且在150℃的油浴上加热的同时借助真空泵在减压下干燥1小时。在氮气气氛下将64mg的茂金属化合物a放入另外准备的100ml的双颈烧瓶中,并且将化合物溶解在13.4ml的脱水甲苯中。在室温下,将8.6ml的由albemarle corporation制造的20%甲基铝氧烷/甲苯溶液添加至茂金属化合物a的甲苯溶液中,接着搅拌30分钟。在将容纳有放置在其中的真空干燥的二氧化硅的200ml的双颈烧瓶在40℃的油浴上加热和搅拌的同时,将全部量的茂金属化合物a与甲基铝氧烷的反应产物的甲苯溶液添加至其中。在将整体在40℃下搅拌1小时之后,仍在40℃的加热下通过蒸馏除去甲苯溶剂,由此获得固体催化剂。
[0367]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0368]
使用在以上实施例1的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0369]
即,在将500ml的充分脱水和脱氧的庚烷、57mg的三乙基铝和34ml的氢气在常压下导入具有搅拌和温度控制装置且内部容积为1升的不锈钢制高压釜之后,在搅拌的同时使温度升高至75℃。导入以相对于乙烯为5mol%的比例包含1
‑
丁烯的乙烯直至其分压达到1.4mpa,将0.100g的以上固体催化剂的10ml的庚烷浆料用氩气压入其中,并且在保持乙烯分压为1.4mpa且温度为75℃的同时继续聚合60分钟。
[0370]
结果,形成了23.6g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.05g/10分钟。聚合条件总结在表6中并且聚合结果总结在表7中。
[0371]
(实施例2)
[0372]
除了使用0.100g的在实施例1中获得的固体催化剂和在导入包含10重量%的1
‑
丁烯的乙烯之前在常压下导入68ml的氢气以外,以与实施例1中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0373]
结果,形成了25.2g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.35g/10分钟。聚合条件总结在表6中并且聚合结果总结在表7中。
[0374]
[表6]
[0375]
表6
[0376][0377]
[表7]
[0378]
表7
[0379][0380]
(实施例3)
[0381]
使用在以上实施例1的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0382]
即,在将800ml的充分脱水和脱氧的异丁烷、34mg的三乙基铝、6483ml的在常压下用氮气稀释至浓度为5%的氢气和10ml的1
‑
丁烯在0.6mpa下导入具有搅拌和温度控制装置且内部容积为1.5升的不锈钢制高压釜之后,在搅拌的同时使温度升高至75℃。导入乙烯直至其分压达到1.4mpa,将0.093g的以上固体催化剂用氮气压入其中,并且在保持乙烯分压为1.4mpa且温度为75℃的同时继续聚合60分钟。
[0383]
结果,形成了108.0g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.20g/10分钟。聚
合条件总结在表8中并且聚合结果总结在表9中。
[0384]
(实施例4)
[0385]
(1)固体催化剂的制备
[0386]
除了使用60mg的茂金属化合物b代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0387]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0388]
使用在以上实施例4的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0389]
除了使用0.209g的在实施例4的(1)固体催化剂的制备中获得的固体催化剂、在0.6mpa下导入30ml的1
‑
丁烯并且不导入氢气以外,以与实施例3中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0390]
结果,形成了53.3g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为3.55g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0391]
(实施例5)
[0392]
(1)固体催化剂的制备
[0393]
除了使用61mg的茂金属化合物c代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0394]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0395]
使用在以上实施例5的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0396]
除了使用0.179g的在实施例5的(1)固体催化剂的制备中获得的固体催化剂、在0.6mpa下导入30ml的1
‑
丁烯并且不导入氢气以外,以与实施例3中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0397]
结果,形成了30.4g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为2.11g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0398]
(实施例6)
[0399]
(1)固体催化剂的制备
[0400]
除了使用62mg的茂金属化合物d代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0401]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0402]
使用在以上实施例6的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0403]
除了使用0.062g的在实施例6的(1)固体催化剂的制备中获得的固体催化剂、在0.6mpa下导入70ml的1
‑
丁烯并且在常压下导入540ml的用氮气稀释至浓度为5%的氢气以外,以与实施例3中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0404]
结果,形成了40.0g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.12g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0405]
(实施例7)
[0406]
(1)固体催化剂的制备
[0407]
除了使用62mg的茂金属化合物e代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0408]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0409]
使用在以上实施例7的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0410]
除了使用0.069g的在实施例7的(1)固体催化剂的制备中获得的固体催化剂、在0.6mpa下导入70ml的1
‑
丁烯并且在常压下导入550ml的用氮气稀释至浓度为5%的氢气以外,以与实施例3中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0411]
结果,形成了5.6g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.24g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0412]
(实施例8)
[0413]
(1)固体催化剂的制备
[0414]
除了使用69mg的茂金属化合物f代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0415]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0416]
使用在以上实施例8的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0417]
除了使用0.038g的在实施例8中获得的固体催化剂、在0.6mpa下导入70ml的1
‑
丁烯并且在常压下导入1958ml的用氮气稀释至浓度为5%的氢气以外,以与实施例3中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0418]
结果,形成了92.2g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.10g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0419]
(比较例1)
[0420]
(1)固体催化剂的制备
[0421]
除了使用54mg的茂金属化合物g代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0422]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0423]
使用在以上比较例1的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0424]
除了使用0.108g的在比较例1的(1)固体催化剂的制备中获得的固体催化剂、在0.6mpa下导入10ml的1
‑
丁烯并且在常压下导入6012ml的用氮气稀释至浓度为5%的氢气以外,以与实施例3中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0425]
结果,形成了38.0g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为1.87g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0426]
(比较例2)
[0427]
(1)固体催化剂的制备
[0428]
除了使用51mg的茂金属化合物h代替茂金属化合物a以外,以与(实施例1)的(1)固体催化剂的制备中相同的方式制备固体催化剂。
[0429]
(2)乙烯
‑1‑
丁烯共聚物的生产
[0430]
使用在以上比较例2的(1)固体催化剂的制备中获得的固体催化剂来生产乙烯
‑1‑
丁烯共聚物。
[0431]
除了使用0.200g的在比较例2的(1)固体催化剂的制备中获得的固体催化剂并且在导入包含10重量%的1
‑
丁烯的乙烯之前在常压下导入68ml的氢气以外,以与实施例1中相同的方式生产乙烯
‑1‑
丁烯共聚物。
[0432]
结果,形成了17.0g的乙烯
‑1‑
丁烯共聚物。所得共聚物的mfr为0.54g/10分钟。聚合条件总结在表8中并且聚合结果总结在表9中。
[0433][0434]
[表9]
[0435]
表9
[0436][0437]
4.评价
[0438]
由表7,在其中使用包含根据本发明的茂金属化合物的催化剂的实施例1和2中,所得乙烯系聚合物的g
a
'值和g
b
'值分别为0.94以下和0.67,因此,揭示的是获得了具有其中引入的长支链的乙烯系聚合物。关于g'值的记载,在未获得任何可靠的g
b
'值的情况下记载为
“‑”
。此外,在g'的最小值存在于logm=5与logm=6之间的情况下,将最小值记载在g
b
'的列的括号内。
[0439]
此外,由表9,示出的是,在其中使用包含根据本发明的茂金属化合物的催化剂的实施例3至7中,所得乙烯系聚合物的g
a
'值为0.76至0.89,因此等于或低于比较例1和2的g
a
'值。实施例3至7的乙烯系聚合物的g
b
'值为0.38至0.59并且低于比较例1和2的g
b
'值,因此,揭示的是与在环戊二烯并噻吩的3位不具有取代基的茂金属化合物(比较例1)和具有茚而不是环戊二烯并噻吩的茂金属化合物(比较例2)的情况相比,获得了具有其中引入的更多长支链的乙烯系聚合物。所述乙烯系聚合物为在伸长粘度行为与熔融流动性之间具有优异的平衡并且具有良好的成形加工性的乙烯系聚合物。此外,在实施例8中获得的所得乙烯系聚合物的g
a
'值和g
b
'值等于比较例1和2的值,但是获得了聚合活性非常高且生产性也令人满意的结果。
[0440]
虽然已经详细地并且参考其特定的实施方案描述了本发明,但是对本领域技术人员将显而易见的是,在不偏离其精神和范围的情况下,可以在其中作出各种变化和修改。
[0441]
本技术基于2017年3月3日提交的日本专利申请no.2017
‑
041011,并且将该内容通过参考引入本文中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1