一种对乙酰氨基苯磺酰氯的制备方法与流程
1.本发明属于磺胺类抗菌药物中间体制备技术领域,具体涉及到一种对乙酰氨基苯磺酰氯的制备方法。
背景技术:
2.对乙酰氨基苯磺酰氯属于磺胺类抗菌药物关键中间体,还可以用作分散染料的中间体,用途广泛。对乙酰氨基苯磺酰氯,化学名称为:4
‑
乙酰胺基苯磺酰氯,英文名称:n
‑
acetylsulfanilyl chloride,cas登记号为:121
‑
60
‑
8,化学结构式如下:
[0003][0004]
对乙酰氨基苯磺酰氯的生产目前采用的是传统工艺(详见:国家医药管理总局.全国原料药工艺汇编,p149
‑
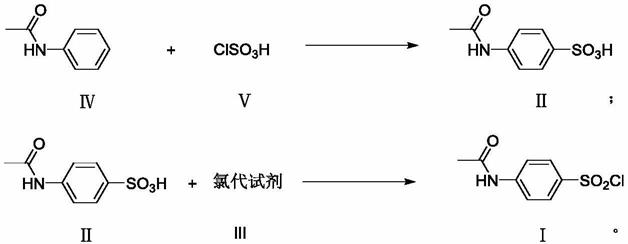
150):以乙酰苯胺与氯磺酸进行磺化反应、制得对乙酰氨基苯磺酸、继续与过量的氯磺酸进行氯化反应得到对乙酰氨基苯磺酰氯,主要反应式如下:
[0005][0006]
该工艺的缺点:1)在氯磺化反应中,由于磺化试剂和氯代试剂均为氯磺酸,但氯磺酸的氯化性能较差,产品收率低,为了得到较好的经济收率,需采用远高于理论用量的氯磺酸,因此氯磺酸用量达6摩尔多,而理论用量只需2摩尔,有4摩尔多的氯磺酸原料未参与反应,因此在后处理过程中,会产生大量的硫酸和盐酸,形成大量高浓度的酸废水,据测算每生产一吨磺胺要产生十多吨废酸水,难以处理,环境污染严重,直接影响企业的经济效益;2)由于氯磺酸用量大大过量,氯磺化反应过程在苯环间位也有一少部份被氯化形成间氯杂质,该杂质在后续的反应及精制等过程中难以去除,造成最终产品中仍有较大残留,hplc检测经常超过0.2%、甚至超过1.0%。
[0007]
近来不少研究者在传统工艺上做了一些改进,如以氯化亚砜为氯代试剂或者在氯化反应过程中加入五氯化磷等,但氯代试剂用量仍较大,产品收率低。产品杂质含量高,废酸液并没有得到根本减少等问题。
[0008]
因此,高质量、高收率、低三废和低成本的对乙酰氨基苯磺酰氯新合成工艺路线开发是非常有必要的。
技术实现要素:
[0009]
本发明所要解决的技术问题在于一种对乙酰氨基苯磺酰氯制备方法,解决质量问
题、工艺收率、三废处理、高成本等缺陷。
[0010]
1.一种对乙酰氨基苯磺酰氯式ⅰ的制备方法,其特征在于以乙酰苯胺式ⅳ与氯磺酸式
ⅴ
在催化剂存在下进行磺化反应,制得对乙酰氨基苯磺酸式ⅱ,再与氯代试剂式ⅲ进行氯化反应得到对乙酰氨基苯磺酰氯式ⅰ;其反应式如下:
[0011][0012]
2.根据权利要求1所述的方法,其特征在于:所述磺化反应中乙酰苯胺式ⅳ与氯磺酸式
ⅴ
的摩尔比为1:(1.0~1.1)。
[0013]
3.根据权利要求1所述的方法,其特征在于:所述磺化反应的催化剂为硫酸铵或硫酸铵。
[0014]
4.根据权利要求1所述的方法,其特征在于:所述氯代试剂式ⅲ采用的为碳酰氯(简称:光气)或双(三氯甲基)碳酸酯(简称:三光气)。
[0015]
5.根据权利要求1和4所述的方法,其特征在于:所述氯化反应中对乙酰氨基苯磺酸式ⅱ与氯代试剂式ⅲ碳酰氯(简称:光气)的摩尔比为1:(1.0~2.0)。
[0016]
6.根据权利要求1和4所述的方法,其特征在于:所述氯化反应中对乙酰氨基苯磺酸式ⅱ与氯代试剂式ⅲ双(三氯甲基)碳酸酯(简称:三光气)的摩尔比为1:(0.34~0.8)。
[0017]
7.根据权利要求1所述的方法,其特征在于:所述氯化反应的催化剂为n,n
‑
二甲基甲酰胺(简称:dmf)或n,n
‑
二甲基乙酰胺(简称:dma)。
[0018]
8.根据权利要求1所述的方法,其特征在于:所述氯化反应的反应温度为40℃~60℃。
[0019]
本发明的优点如下:
[0020]
1.本发明的制备方法所得的对乙酰氨基苯磺酰氯较现有工艺收率高,摩尔收率可高达88.0~95.0%。
[0021]
2.本发明的制备方法所得的对乙酰氨基苯磺酰氯,由于没有使用大大过量的氯磺酸,减少了间氯杂质的产生,最终产品中间氯杂质极少,同时其他杂质小于0.1%,产品纯度在99.0%以上。
[0022]
3.本发明的制备方法与现有技术中的方法相比,由于氯磺酸使用量为理论用量,从而避免了大量废酸水的产生,废酸水量不到原工艺的10%,从源头上根本解决了环保处理问题。
[0023]
4.本发明的制备方法与现有技术中的方法相比,氯代试剂由原来的氯磺酸更换为碳酰氯或双(三氯甲基)碳酸酯,它们在反应中仅会产生氯化氢气体及二氧化碳气体,氯化氢气体可以用水吸收制备盐酸,而二氧化碳可以直接排放,因此环保处理相对较容易,大幅度减轻环保处理压力。
[0024]
5.本发明的制备方法与现有技术中的方法相比工艺操作简单,对生产设备无特殊
要求,减轻了对设备的腐蚀程度,适于工业化生产。
[0025]
本发明所用原料乙酰苯胺容易得到,也可通过现有文献公开的方法制得(樊能延编著.有机合成事典p183),具体工艺如下:以苯胺为原料、和醋酐进行乙酰化反应制得乙酰苯胺,其反应式如下:
[0026][0027]
本发明所用原料氯磺酸容易得到,本发明所用原料氯代试剂碳酰氯(简称:光气)或双(三氯甲基)碳酸酯(简称:三光气)也是容易购买的。
[0028]
通过实施例进一步说明和解释本发明的制备方法,但不限制本发明的范围。
具体实施方式
[0029]
实施例1对乙酰氨基苯磺酸式ⅱ的制备
[0030]
分别将乙酰苯胺式ⅳ62.2g(0.46mol)、硫酸铵3.0g加入到反应瓶中,冷却至0℃~5℃,再加入氯磺酸式
ⅴ
53.6g(0.46mol),升温至40℃~50℃搅拌反应2小时,反应过程中有氯化氢气体产生,反应结束,冷却,过滤,得到白色固体对乙酰氨基苯磺酸式ⅱ98.2g,收率99.2%。
[0031]
实施例2对乙酰氨基苯磺酸式ⅱ的制备
[0032]
分别将乙酰苯胺式ⅳ202.7g(1.50mol)、硫酸铵6.0g加入到反应瓶中,冷却至0℃~5℃,再加入氯磺酸式
ⅴ
209.7g(1.8mol),升温至35℃~45℃搅拌反应2小时,反应过程中有氯化氢气体产生,反应结束,冷却,过滤,得到白色固体对乙酰氨基苯磺酸式ⅱ319.0g,收率98.8%。
[0033]
实施例3对乙酰氨基苯磺酰氯式ⅰ的制备
[0034]
分别将二氯甲烷271.2g、对乙酰氨基苯磺酸式ⅱ90.4g(0.42mol)、n,n
‑
二甲基甲酰胺(简称:dmf)3.1g加入到反应瓶中,滴加碳酰氯的二氯甲烷溶液(事先将碳酰氯45.5g(0.46mol)通入二氯甲烷45.5g中),滴加完后升温至38℃~45℃反应2小时,反应过程中有氯化氢和二氧化碳气体产生,冷却至0℃~10℃,过滤,得对乙酰氨基苯磺酰氯式ⅰ90.8g,收率92.5%。
[0035]
实施例4对乙酰氨基苯磺酰氯式ⅰ的制备
[0036]
分别将三氯甲烷241.4g、对乙酰氨基苯磺酸式ⅱ53.8g(0.0.25mol)、n,n
‑
二甲基乙酰胺(简称:dma)2.5g加入到反应瓶中,滴加碳酰氯的三氯甲烷溶液(事先将碳酰氯37.6g(0.38mol)通入三氯甲烷37.6g中),滴加完后升温至38℃~45℃反应2小时,反应过程中有氯化氢和二氧化碳气体产生,冷却至0℃~10℃,过滤,得对乙酰氨基苯磺酰氯式ⅰ55.1g,收率94.3%。
[0037]
实施例5对乙酰氨基苯磺酰氯式ⅰ的制备
[0038]
分别将双(三氯甲基)碳酸酯29.7g(0.10mol)、三氯甲烷29.7g加入到反应瓶中,搅拌溶解备用。
[0039]
分别将三氯甲烷180.9g、对乙酰氨基苯磺酸式ⅱ60.3g(0.28mol)、n,n
‑
二甲基甲酰胺(简称:dmf)1.8g加入到另一反应瓶中,滴加双(三氯甲基)碳酸酯的二氯甲烷溶液,滴加完后升温至38℃~45℃反应2小时,反应过程中有氯化氢和二氧化碳气体产生,冷却至0
℃~10℃,过滤,得对乙酰氨基苯磺酰氯式ⅰ62.5g,收率95.5%。
[0040]
实施例6对乙酰氨基苯磺酰氯式ⅰ的制备
[0041]
分别将双(三氯甲基)碳酸酯89.0g(0.30mol)、四氯化碳89.0g加入到反应瓶中,搅拌溶解备用。
[0042]
分别将乙酰苯胺式ⅳ75.7g(0.56mol)、氯化铵3.0g加入到另一反应瓶中,冷却至0℃~5℃,再加入氯磺酸式ⅲ65.3g(0.56mol),升温至40℃~50℃搅拌反应2小时,反应过程中有氯化氢气体产生,反应结束,冷却,直接进行下一步反应。
[0043]
在上述反应体系中,分别加入四氯化碳262g、n,n
‑
二甲基甲酰胺(简称:dmf)2.0g,滴加双(三氯甲基)碳酸酯的四氯化碳溶液,滴加完后升温至38℃~45℃反应2小时,反应过程中有氯化氢和二氧化碳气体产生,冷却至0℃~10℃,过滤,得对乙酰氨基苯磺酰氯式ⅰ123.0g,收率94.0%
[0044]
实施例7对乙酰氨基苯磺酰氯式ⅰ的制备(对比实验)
[0045]
(参见国家医药管理总局.全国原料药工艺汇编,p149
‑
150)
[0046]
将乙酰苯胺式ⅳ40.0g(0.30mol)在20℃均匀加入氯磺酸式
ⅴ
210.4g(1.81mol),在48放置8小时后,约20℃以下,加入饮用水分解过剩的氯磺酸,温度不超过28℃,冷却,过滤,洗涤滤饼至ph=3~4,得对乙酰氨基苯磺酰氯式ⅰ57.8g,收率82.5%。
[0047]
前面已经详述了本发明,包括其优选的实施方案。但是应当明白,考虑到本发明公开的内容,本领域技术人员可在所述权利要求书的精神范围内对本发明进行改变和/或改进,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1