一种维生素B1中间体的绿色合成方法与流程
一种维生素b1中间体的绿色合成方法
技术领域
1.本发明涉及维生素b1的生产技术领域,具体涉及一种维生素b1中间体3
‑
氯
‑5‑
羟基
‑2‑
戊酮的绿色合成方法。
背景技术:
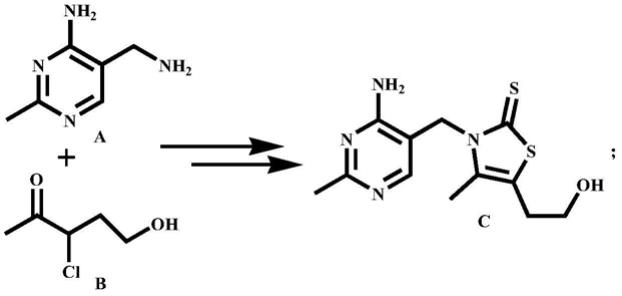
2.维生素b1又名硫胺素,作为第一个被发现的水溶性维生素和合成路线最长的维生素更是受到人们的普遍关注,用以解决神经炎、心肌炎、消化不良等疾病的辅助治疗及脚气病的防治。其关键中间体硫代硫胺(c)工业上是由双胺嘧啶(a)和3
‑
氯
‑5‑
羟基
‑2‑
戊酮(b)作为起始原料通过一系列反应制备而成,如下式所示:
[0003][0004]3‑
氯
‑5‑
羟基
‑2‑
戊酮是合成硫代硫胺的重要中间体,关于3
‑
氯
‑5‑
羟基
‑2‑
戊酮的合成,现有工艺主要以α
‑
乙酰基
‑
γ
‑
丁内酯为起始原料,在氯气或磺酰氯氯化试剂的作用下,发生取代反应生成α
‑
氯代乙酰基
‑
γ
‑
丁内酯,而α
‑
氯代乙酰基
‑
γ
‑
丁内酯进一步在稀盐酸或稀硫酸的作用下脱羰生成3
‑
氯
‑5‑
羟基
‑2‑
戊酮。
[0005]
例如:以α
‑
乙酰基
‑
γ
‑
丁内酯为起始原料,使用氯气作为氯化试剂制备3
‑
氯
‑5‑
羟基
‑2‑
戊酮的工艺,反应如下所示:
[0006][0007]
在制备3
‑
氯
‑5‑
羟基
‑2‑
戊酮的过程中,此工艺有用到强烈刺激性的剧毒气体氯气,且反应过程中还会生成当量的hcl,无论从安全性上,还是从原子经济性上,都不符合绿色合成的宗旨。
[0008]
又如:以α
‑
乙酰基
‑
γ
‑
丁内酯为原料,经过氨化、氯代、缩合、氧化反应而合成4
‑
甲基
‑5‑
(2
‑
羟乙基)
‑
噻唑;其中原料α
‑
乙酰基
‑
γ
‑
丁内酯与氯气在紫外光的照射下反应,水解得3
‑
氯
‑5‑
羟基
‑2‑
戊酮,该制备方法不仅使用具有强烈刺激性的剧毒气体氯气,且反应需要额外的能量源。
[0009]
又如:以α
‑
乙酰基
‑
γ
‑
丁内酯为起始原料,使用磺酰氯作为氯化试剂,经稀硫酸水解脱羧制备3
‑
氯
‑5‑
羟基
‑2‑
戊酮。该制备方法会产生大量有毒有害的二氧化硫、盐酸等废
气,对人体、环境的危害都非常大,且后处理比较麻烦,二氧化硫残留对后一步反应的影响较大,并且氯代试剂的用量难以控制,易产生多氯等副产物。
技术实现要素:
[0010]
本发明的目的是克服现有技术的不足,提供一种对环境更友好、设备腐蚀性小且兼具较高收率的3
‑
氯
‑5‑
羟基
‑2‑
戊酮(维生素b1中间体)的绿色合成方法。
[0011]
本发明同时还提供了一种α
‑
羟基乙酰基
‑
γ
‑
丁内酯(可作为维生素b1中间体或3
‑
氯
‑5‑
羟基
‑2‑
戊酮中间体)的合成方法。
[0012]
为达到上述目的,本发明采用的一种技术方案是:一种式(ⅲ)所示的化合物(3
‑
氯
‑5‑
羟基
‑2‑
戊酮)的合成方法,该合成方法包括:在催化剂存在下使式(ⅰ)所示的化合物(α
‑
乙酰基
‑
γ
‑
丁内酯)在有氧条件下、在溶剂中反应,生成式(ⅱ)所示的化合物(α
‑
羟基乙酰基
‑
γ
‑
丁内酯),然后向反应体系中通入氯化氢,依次进行取代反应和水解脱羧反应,生成式(ⅲ)所示的化合物;其中,所述催化剂包括铜盐类路易斯酸;
[0013][0014]
根据本发明的一些优选且具体的方面,所述铜盐类路易斯酸为一价铜盐和/或二价铜盐。
[0015]
根据本发明的一些优选方面,所述铜盐类路易斯酸的酸根为选自氯离子、醋酸根、硫酸根和硝酸根中的一种或多种的组合。
[0016]
根据本发明的一些优选方面,所述铜盐类路易斯酸为选自氯化铜、氯化亚铜、醋酸铜、硫酸铜、硫酸亚铜和硝酸铜中的一种或多种的组合。
[0017]
根据本发明的一些优选方面,所述催化剂还包括除所述铜盐类路易斯酸以外的其他路易斯酸。进一步地,所述铜盐类路易斯酸与所述除所述铜盐类路易斯酸以外的其他路易斯酸的投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1。在本发明的一些实施方式中,所述铜盐类路易斯酸与所述除所述铜盐类路易斯酸以外的其他路易斯酸的投料摩尔比可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等。
[0018]
根据本发明的一些具体且优选的方面,所述除所述铜盐类路易斯酸以外的其他路易斯酸为选自铁盐、钯盐、锰盐、锌盐、钴盐、钌盐、铑盐和铝盐中的一种或多种的组合。
[0019]
根据本发明的一些优选方面,所述除所述铜盐类路易斯酸以外的其他路易斯酸为选自氯化锌、溴化锌、氯化锰、氯化钴、氯化铁、三氯化铝、醋酸锌、醋酸锰、醋酸钴、三氟乙酸钯和三氯化钌中的一种或多种的组合。
[0020]
根据本发明的一些优选且具体的方面,所述催化剂为选自如下(a)
‑
(i)组合中的一种或多种:
[0021]
(a)氯化亚铜和氯化锰,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、
0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0022]
(b)氯化亚铜和醋酸钴,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0023]
(c)氯化亚铜和三氯化钌,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0024]
(d)醋酸铜和氯化钴,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0025]
(e)硫酸铜和醋酸锌,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0026]
(f)硫酸亚铜和三氟乙酸钯,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0027]
(g)氯化铜和醋酸锰,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0028]
(h)硝酸铜和醋酸钴,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等;
[0029]
(i)硫酸铜和三氟乙酸钯,投料摩尔比为1∶0.001
‑
1,更优选为1∶0.01
‑
1,例如可以为1∶0.900
‑
1、1∶0.800
‑
0.899、1∶0.700
‑
0.799、0.600
‑
0.699、0.500
‑
0.599、0.400
‑
0.499、0.300
‑
0.399、0.200
‑
0.299、0.100
‑
0.199、0.010
‑
0.099等。
[0030]
根据本发明的一些优选方面,以摩尔百分含量计,所述催化剂的摩尔添加量为式(ⅰ)所示的化合物的1.5
‑
6.0mol%。
[0031]
根据本发明的一些优选方面,所述有氧条件为通过向反应体系中通入氧气和/或空气形成,空气或氧气是最绿色的氧化剂,本发明采用空气或氧气作为终端氧化剂能够满足绿色化工和可持续发展化学的要求,且反应效果好。
[0032]
根据本发明的一些优选方面,使生成式(ⅱ)所示的化合物的所述反应在反应压力为0.1
‑
3.0mpa下进行,进一步地可以为0.5
‑
1.5mpa。
[0033]
根据本发明的一些优选方面,使生成式(ⅱ)所示的化合物的所述反应在反应温度为80
‑
110℃下进行,进一步地可以为在反应温度为85
‑
100℃下进行。
[0034]
根据本发明的一个优选方面,所述溶剂为水,本发明反应过程中采用水作为氧化反应溶剂,工艺路线会更加地安全环保,相对于传统有机溶剂,无爆炸风险,方法安全性高,更适用于工业生产,且反应效果好。
[0035]
根据本发明的一些优选方面,所述式(ⅰ)所示的化合物与所述溶剂的投料质量比为1∶5
‑
8。
[0036]
根据本发明的一些优选方面,使所述取代反应在反应温度为10
‑
40℃下进行,进一步地可以为在反应温度为20
‑
30℃下进行。
[0037]
根据本发明的一些优选方面,使所述水解脱羧反应在反应温度为60
‑
80℃下进行,进一步地可以在例如反应温度为60
‑
65℃、65
‑
70℃、70
‑
75℃、75
‑
80℃下进行。
[0038]
根据本发明的一些优选方面,控制所述取代反应的反应时间为2
‑
4h。
[0039]
根据本发明的一些优选方面,控制所述水解脱羧反应的反应时间为1
‑
4h。
[0040]
根据本发明的一些优选方面,在进行所述取代反应、所述水解脱羧反应的过程中,通过控制氯化氢的通入量以使反应体系的起始反应压力为0.1
‑
0.6mpa,进一步地可以为0.2
‑
0.5mpa。
[0041]
根据本发明,本发明合成式(ⅲ)所示的化合物的方法能够采用一锅法进行,具有步骤经济的优点,且减少反应后处理步骤,操作更简便,降低了生产成本,且反应效果好。
[0042]
根据本发明的一些优选且具体的方面,所述合成方法采用如下实施方式:
[0043]
将式(ⅰ)所示的化合物、催化剂和水加入反应釜中,采用氧气置换多次,并向反应釜内通入氧气直至釜内压力为0.1
‑
3.0mpa且一直维持,将釜内温度升至80
‑
110℃,反应8
‑
16h,反应结束后,降低釜内温度至10
‑
40℃,将釜内氧气排空,通入氯化氢,当反应体系的起始反应压力为0.1
‑
0.6mpa时,停止通入,反应2
‑
4h后,升温至60
‑
80℃,反应1
‑
4h,生成所述式(ⅲ)所示的化合物。
[0044]
根据本发明的一些具体方面,通过核磁内标法测定反应后反应混合液中式(ⅲ)所示的化合物的含量,内标物可以为二溴甲烷。
[0045]
本发明提供的又一技术方案:采用上述所述的合成方法制备式(ⅱ)所示的化合物;其反应如下所示:
[0046][0047]
本发明提供的又一技术方案:一种维生素b1的制备方法,所述制备方法包括上述所述的式(ⅲ)所示的化合物的合成方法。
[0048]
由于上述技术方案运用,本发明与现有技术相比具有下列优点:
[0049]
本发明基于现有技术中制备3
‑
氯
‑5‑
羟基
‑2‑
戊酮(式(ⅲ)所示的化合物)存在的均需使用氯气或磺酰氯氯化试剂等本身具有高腐蚀、高毒性或其反应产物中会产生大量有毒有害废气的缺陷,创新地提供了一种新的且兼具对环境更友好、设备腐蚀性小、能够获得较高收率的3
‑
氯
‑5‑
羟基
‑2‑
戊酮(式(ⅲ)所示的化合物)的合成方法,该方法采用使式(ⅰ)所示的化合物在特定的催化剂下、在有氧条件下、在溶剂中反应,高收率的生成式(ⅱ)所示的化合物,然后使式(ⅱ)所示的化合物在氯化氢作用下依次进行取代反应和水解脱羧反应,最终制成式(ⅲ)所示的化合物,该路线中,不仅避免了使用氯气或磺酰氯氯化试剂等环境不友好物质,而且氧气或空气所含氧气作为氧化反应的终端氧化剂,氧化剂绿色可靠,尤其是还能够获得优异的收率,且该反应路线还能够实现一锅法制备3
‑
氯
‑5‑
羟基
‑2‑
戊酮,使操作更简单,生产成本更低,有利于工业化应用。
具体实施方式
[0050]
以下结合具体实施例对上述方案做进一步说明;应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制;实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
[0051]
下述实施例中未作特殊说明,所有原料可以来自于商购或通过本领域的常规方法制备而得。
[0052]
实施例1
[0053]
取220g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),0.618g氯化亚铜,0.786g氯化锰,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力约为1.36mpa,且一直维持釜内压力约为1.36mpa,将釜内温度升至85℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.48mpa后,停止通入,在搅拌下继续反应2h。然后缓慢升温至70℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为91.25%。
[0054]
实施例2
[0055]
取220g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),0.618g氯化亚铜,1.105g醋酸钴,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力约为1.36mpa,且一直维持釜内压力约为1.36mpa,将釜内温度升至85℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.32mpa后,停止通入,在搅拌下继续反应2h。然后缓慢升温至70℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为89.36%。
[0056]
实施例3
[0057]
取300g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),0.618g氯化亚铜,1.295g三氯化钌,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.85mpa,且一直维持釜内压力约为0.85mpa,将釜内温度升至85℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.32mpa后,停止通入,在搅拌下继续反应2h。然后缓慢升温至70℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为72.54%。
[0058]
实施例4
[0059]
取200g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.621g醋酸铜,0.730g氯化钴,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.78mpa,且一直维持釜内压力约为0.78mpa,将釜内温度升至95℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排
空,随后通入hcl至釜内起始反应压力为0.21mpa后,停止通入,在搅拌下继续反应2h。然后缓慢升温至60℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为88.47%。
[0060]
实施例5
[0061]
取200g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.122g醋酸铜,0.730g氯化钴,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.78mpa,且一直维持釜内压力约为0.78mpa,将釜内温度升至95℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.21mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至60℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为85.63%。
[0062]
实施例6
[0063]
取240g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.146g硫酸铜,1.557g三氟乙酸钯,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为1.73mpa,且一直维持釜内压力约为1.73mpa,将釜内温度升至100℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.37mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至75℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为79.61%。
[0064]
实施例7
[0065]
取220g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.146g硫酸铜,0.859g醋酸锌,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为1.91mpa,且一直维持釜内压力约为1.91mpa,将釜内温度升至100℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.42mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至75℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为77.46%。
[0066]
实施例8
[0067]
取300g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.951g硫酸亚铜,2.906g三氟乙酸钯,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.96mpa,且一直维持釜内压力约为0.96mpa,将釜内温度升至90℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.25mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至80℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊
酮的收率为79.82%。
[0068]
实施例9
[0069]
取320g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.049g氯化铜,0.972g醋酸锰,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为2.55mpa,且一直维持釜内压力约为2.55mpa,将釜内温度升至90℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.34mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至80℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为80.74%。
[0070]
实施例10
[0071]
取320g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.049g氯化铜,0.972g醋酸锰,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,向高压釜内通入空气增压直至釜内压力为1.25mpa,且一直维持釜内压力约为1.25mpa,将釜内温度升至90℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内压力排空,随后通入hcl至釜内起始反应压力为0.34mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至80℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为77.03%。
[0072]
实施例11
[0073]
取320g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.049g氯化铜,0.972g醋酸锰,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为1.25mpa,且一直维持釜内压力约为2.55mpa,将釜内温度升至90℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.34mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至80℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为89.86%。
[0074]
实施例12
[0075]
取280g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.757g硝酸铜,0.829g醋酸钴,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.67mpa,且一直维持釜内压力约为0.67mpa,将釜内温度升至85℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.28mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至75℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为90.08%。
[0076]
实施例13
[0077]
取280g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.757g硝酸铜,加入到带有搅
拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.67mpa,且一直维持釜内压力约为0.67mpa,将釜内温度升至85℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.28mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至75℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为82.14%。
[0078]
实施例14
[0079]
取300g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.951g硫酸亚铜,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.96mpa,且一直维持釜内压力约为0.96mpa,将釜内温度升至90℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.25mpa后,停止通入,在搅拌下继续反应3h。然后缓慢升温至80℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为73.57%。
[0080]
实施例15
[0081]
取200g h2o,40gα
‑
乙酰基
‑
γ
‑
丁内酯(0.3122mol),1.621g醋酸铜,加入到带有搅拌和冷却回流装置的500ml衬四氟乙烯的密闭高压釜内,氧气置换3次,然后向高压釜内通入氧气至釜内压力为0.78mpa,且一直维持釜内压力约为0.78mpa,将釜内温度升至95℃,在搅拌下反应10h。反应结束后,釜内温度缓慢降至25℃后,将釜内氧气排空,随后通入hcl至釜内起始反应压力为0.21mpa后,停止通入,在搅拌下继续反应2h。然后缓慢升温至60℃,在搅拌下继续反应2h,反应结束后,冷却至室温,将高压釜排气口连接尾气吸收装置后打开,排空后取样,以核磁内标法(内标为二溴甲烷)确定3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为83.46%。
[0082]
对比例1
[0083]
基本同实施例1,其区别仅在于:不添加氯化亚铜,相应调整氯化锰的添加量为1.560g(保持催化剂为同等摩尔添加量),3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为63.07%。
[0084]
对比例2
[0085]
基本同实施例1,其区别仅在于:不添加催化剂氯化亚铜和氯化锰,更换催化剂为氯化钴,氯化钴的添加量为1.610g(保持催化剂为同等摩尔添加量),3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为61.23%。
[0086]
对比例3
[0087]
基本同实施例1,其区别仅在于:不添加催化剂氯化亚铜和氯化锰,更换催化剂为氯化锌,氯化锌的添加量为1.690g(保持催化剂为同等摩尔添加量),并未获得3
‑
氯
‑
5羟基
‑2‑
戊酮。
[0088]
对比例4
[0089]
基本同实施例1,其区别仅在于:不添加催化剂氯化亚铜和氯化锰,更换催化剂为三氯化铈,三氯化铈的添加量为3.056g(保持催化剂为同等摩尔添加量),3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为47.56%。
[0090]
对比例5
[0091]
基本同实施例1,其区别仅在于:不添加催化剂氯化亚铜和氯化锰,更换催化剂为三氟乙酸钯,三氟乙酸钯的添加量为4.122g(保持催化剂为同等摩尔添加量),3
‑
氯
‑
5羟基
‑2‑
戊酮的收率为30.72%。
[0092]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
[0093]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0094]
本发明的工作得到来自“2020年度中国博士后创新人才支持计划”的基金支持,基金资助编号:bx20200306。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1