一种Avacopan及其中间体的制备方法与流程
一种avacopan及其中间体的制备方法
技术领域
1.本发明涉及有机合成技术领域,具体涉及一种avacopan及其中间体的制备方法。
背景技术:
2.补体系统在清除感染源、外源抗原、被病毒感染的细胞和肿瘤细胞的免疫应答中起着非常重要的作用。但补体系统的不适当或过度激活可能会导致严重的炎症和组织破坏,这些结果甚至可以危及生命。这些后果在临床上表现为各种疾病,包括移植排斥、肾炎、病理性炎症和自身免疫性疾病等(补体系统在创伤和感染中的作用,《国际外科学杂志》,1993年第1期17-18)。
3.补体途径的激活会产生补体蛋白的生物活性片段。例如,c3a、c4a和c5a过敏反应蛋白和c5b-9膜攻击复合物(mac),这些生物活性片段均通过影响白细胞的趋化性从而介导炎症反应,激活中性粒细胞、血小板、肥大细胞和内皮细胞,增加血管通透性,促使细胞溶解等一系列炎症损伤。
4.补体c5a是补体系统中最有效的促炎介质之一,由两个多肽链α和β组成,其分子量分别约为115kd和75kd。过敏性c5a肽在引起炎症反应方面的效力是c3a的100倍。c5a是c5的激活形式,其存在于人体血清中,含量约为80μg/ml,除了其过敏反应特性外,c5a还能诱导中性粒细胞、嗜碱性粒细胞和单核细胞的趋化迁移。已有研究表明,c5a的过敏性和趋化作用是通过与c5a受体的相互作用来介导的。人c5a受体(c5ar)是一种52kd的位于细胞膜上的g蛋白偶联受体,在中性粒细胞、单核细胞、嗜碱性粒细胞、嗜酸性粒细胞、肝细胞、肺平滑肌和内皮细胞以及肾小球等细胞或组织上表达,c5ar的配体结合位点是高度复杂的,由至少两个可分离的结合域组成:一个结合c5a氨基端(aa1-20)和二硫键(aa21-61),而第二个结合c5a羧基端(aa62-74),因此,c5a的肽类受体拮抗剂获取难度大。
5.直到chemocentryx公司研发的选择性补体c5a非肽类受体抑制剂avacopan,可抑制c5a诱导的免疫细胞活化,其主要适应症为:1、抗中性粒细胞胞浆抗体(anca)相关性血管炎(aav);2、c3肾小球病(c3g);3、化脓性汗腺炎(hs)。
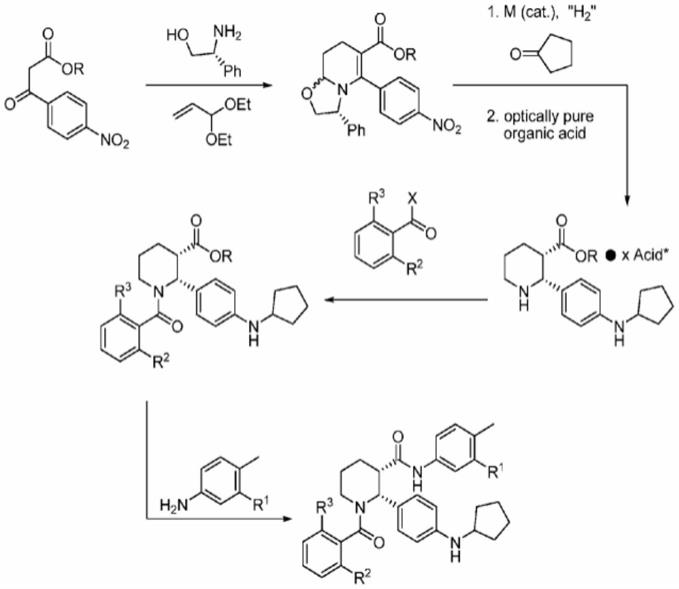
6.美国专利us2016090357a1公开了三种avacopan的制备方法,第一种制备方法如下:
[0007][0008]
第二种制备方法如下:
[0009][0010]
第三种制备方法如下:
[0011][0012]
上述的3种方法中,第一种方法和第二种方法类似,只是反应顺序有所区别,两种方法都涉及到了手性辅基,原子经济性比较低;且钯催化的氢化反应不是非常具有选择性,需要拆分富集手性异构体,这会导致目标产物纯度和收率的降低,因此,两种制备方法的成本都较高。第三种方法,涉及到钯催化的suzuki反应,铂催化的氢化反应,原子经济性低,不适合工业化生产。
技术实现要素:
[0013]
本发明为解决上述问题,提供了一种选择性高、收率高、成本更低的avacopan的制备方法。
[0014]
为实现上述目的,本发明采用的技术方案如下:
[0015]
本发明公开了一种avacopan中间体的制备方法,所述式(ⅳ)所示化合物由式(
ⅴ
)所示化合物和异丙胺或其盐发生转氨反应得到,所述转氨反应是在ω-转氨酶和磷酸吡哆醛的催化下进行。
[0016][0017]
优选地,上述反应的反应温度为10-70℃。
[0018]
优选地,上述式(
ⅴ
)所示化合物和异丙胺或其盐的摩尔比为1:1-1:4。
[0019]
优选地,上述式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为100:1-200:1。
[0020]
优选地,上述式(
ⅴ
)所示化合物和ω-转氨酶的质量比为20:1-100:1。
[0021]
本发明还公开了式(ⅲ)所示化合物的制备方法,由上述式(ⅳ)所示化合物经硼烷四氢呋喃溶液还原得到。
[0022][0023]
优选地,上述式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为1:1-1:5。
[0024]
本发明还公开了式(ⅱ)所示化合物的制备方法,由上述式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯发生酰化反应得到。
[0025]
优选地,上述式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩尔比为1:1-1:4。
[0026][0027]
本发明还公开了式(ⅰ)所示化合物的制备方法,由所述式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺发生胺酯交换反应得到。
[0028][0029]
优选地,上述式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺的摩尔比为1:1-1:4。
[0030]
本发明还公开了一种avacopan的制备方法,由上述式(ⅰ)所示化合物和环戊酮通过还原胺化反应制备得到,所述还原胺化反应是在钯碳的催化和氢气的参与下进行。
[0031][0032]
优选地,上述式(ⅰ)所示化合物和环戊酮的摩尔比为1:1-1:4。
[0033]
优选地,上述式(ⅰ)所示化合物和钯碳的摩尔比为100:1-10:1。
[0034]
相对于现有技术,本发明具有以下有益效果:
[0035]
ω-转氨酶和磷酸吡哆醛可以催化上述式(
ⅴ
)所示化合物2-(4-硝基苯甲酰)戊酸二乙酯的不对称转氨化反应,选择性非常高,几乎没有非对应异构体生成,ee%(对映体超量)》98.0%,即得到的上述式(ⅳ)所示化合物(2r,3s)-2-(4-硝基苯基)-6-氧代哌啶-3-羧酸乙酯的含量大于99%,而且反应产率大于80%,极大程度上解决了现有技术中反应选择性不好,非对应异构体难以除去或者纯化成本高昂,原子经济性低,不适合工业化生产等问题。
具体实施方式
[0036]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0037]
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。
[0038]
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本文中使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同意义。
[0039]
值得说明的是,本发明中使用的原料均为普通市售产品,对其来源不做具体限定,原料来源如表1所示。
[0040]
表1
[0041][0042]
实施例1-5 一种avacopan中间体式(ⅳ)所示化合物的制备方法
[0043]
基础实施例:所述式(ⅳ)所示化合物由式(
ⅴ
)所示化合物和异丙胺或其盐发生转氨反应得到,所述转氨反应是在ω-转氨酶和磷酸吡哆醛的催化下进行。
[0044][0045]
在250ml的三口瓶中加入式(
ⅴ
)所示化合物2-(4-硝基苯甲酰)戊酸二乙酯(33.7g,0.1mol),5%dmso(1l),搅拌溶解;加入二异丙胺盐(19.0g,0.2mol)、na2hpo4和na2hpo4的缓冲液(ph=7.5,150ml);控制反应温度为60-70℃,加入ω-转氨酶(1g)和磷酸吡哆醛(0.247g,1mmol),10℃搅拌反应2天。用200ml乙酸乙酯萃取,重复萃取两次;有机相浓缩至约200ml,升温至70-80℃,滴加正庚烷(200ml);自然冷却至20-30℃,过滤,干燥后得式(ⅳ)所示化合物(2r,3s)-2-(4-硝基苯基)-6-氧代哌啶-3-羧酸乙酯(24.8g)。
[0046]
实施例2
[0047]
本实施例与实施例1的不同在于,式(
ⅴ
)所示化合物和二异丙胺盐的摩尔比为1:4,式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为200:1,式(
ⅴ
)所示化合物和ω-转氨酶的质量比为100:1,反应温度为30-40℃,其余和实施例1相同。
[0048]
实施例3
[0049]
本实施例与实施例1的不同在于,式(
ⅴ
)所示化合物和二异丙胺盐的摩尔比为1:1,式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为150:1,式(
ⅴ
)所示化合物和ω-转氨酶的质量比为20:1,反应温度为10-20℃,其余和实施例1相同。
[0050]
实施例4
[0051]
本实施例与实施例1的不同在于,式(
ⅴ
)所示化合物和二异丙胺盐的摩尔比为1:2,式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为120:1,式(
ⅴ
)所示化合物和ω-转氨酶的质量比为70:1,反应温度为40-50℃,其余和实施例1相同。
[0052]
实施例5
[0053]
本实施例与实施例1的不同在于,式(
ⅴ
)所示化合物和二异丙胺盐的摩尔比为1:3,式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为20:1,式(
ⅴ
)所示化合物和ω-转氨酶的质量比为50:1,反应温度为20-30℃,其余和实施例1相同。
[0054]
对比例1
[0055]
本对比例与实施例1的不同在于,式(
ⅴ
)所示化合物和二异丙胺盐的摩尔比为1:5,式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为60:1,其余和实施例1相同。
[0056]
对比例2
[0057]
本对比例与实施例1的不同在于,式(
ⅴ
)所示化合物和ω-转氨酶的质量比为200:1,反应温度为2-8℃,其余和实施例1相同。
[0058]
对比例3
[0059]
本对比例与实施例1的不同在于,式(
ⅴ
)所示化合物和二异丙胺盐的摩尔比为1:5,反应温度为2-8℃,其余和实施例1相同。
[0060]
对比例4
[0061]
本对比例与实施例1的不同在于,式(
ⅴ
)所示化合物和ω-转氨酶的质量比为200:1,式(
ⅴ
)所示化合物和磷酸吡哆醛的摩尔比为50:1,其余和实施例1相同。
[0062]
对比例5
[0063]
本对比例与实施例1的不同在于,反应温度为75-80℃,其余和实施例1相同。
[0064]
按照上述实施例1-5和对比例1-5制得式(ⅳ)所示化合物,其纯度、ee%和收率如表2所示。
[0065]
表2
[0066] 产物纯度ee%收率实施例199.2%98.5%85%实施例299.3%98.6%86%实施例399.3%98.7%87%实施例499.4%98.6%88%实施例599.0%99.0%87%对比例194.2%88.7%76%对比例294.3%88.6%76%对比例394.3%88.8%75%对比例494.4%88.5%78%对比例594.0%89.0%77%
[0067]
上述结果表明,实施例1-5中各原料组分的比例在本发明的保护范围内,制得的产物纯度均在99%以上,ee%均在98.5%以上,证明非对应异构体的量极少,且收率均在85%以上。而对比例1-5中部分原料组分的比例不在本发明的保护范围内,其产物纯度低,非对
应异构体的含量高且产率低。
[0068]
实施例6-9 一种avacopan中间体式(ⅲ)所示化合物和式(ⅱ)所示化合物的制备方法。
[0069]
基础实施例:式(ⅲ)所示化合物的制备方法,由式(ⅳ)所示化合物经硼烷四氢呋喃溶液还原得到。
[0070][0071]
式(ⅱ)所示化合物的制备方法,由式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯发生酰化反应得到。
[0072][0073]
实施例6
[0074]
在250ml的三口瓶中加入式(ⅳ)所示化合物(14.6g,0.05mol)和150ml四氢呋喃(thf),搅拌溶解;0-20℃下滴加1m硼烷四氢呋喃溶液150ml;滴加完毕后,20-30℃,搅拌反应12小时。0-15℃,滴加1m盐酸300ml淬灭反应;20~30℃,搅拌反应24小时,得到式(ⅲ)所示化合物。用10%碳酸钠溶液调节ph至9-11,10-30℃下滴加2-氟-6-甲基苯甲酰氯(17.2g,0.1mol),滴加时间1h;20-30℃反应2-4h;反应液低于40℃减压浓缩除去大部分thf,使用100ml二氯甲烷萃取,重复一次萃取;合并有机相,减压浓缩至不滴;加入醋酸异丙酯(120ml),80-90℃搅拌0.5h(固体全部溶解);缓慢冷却至10-20℃,固体逐渐析出,过滤,50-60℃减压干燥,得式(ⅱ)所示化合物。
[0075]
实施例7
[0076]
本实施例与实施例6的不同在于,式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为1:1,式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩尔比为1:4,其余和实施例6相同。
[0077]
实施例8
[0078]
本实施例与实施例6的不同在于,式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为1:5,式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩尔比为1:1,其余和实施例6相同。
[0079]
实施例9
[0080]
本实施例与实施例6的不同在于,式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为1:4,式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩尔比为1:3,其余和实施例6相同。
[0081]
对比例6
[0082]
本对比例与实施例6的不同在于,式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为1:10,其余和实施例6相同。
[0083]
对比例7
[0084]
本对比例与实施例6的不同在于,式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩
尔比为1:2,其余和实施例6相同。
[0085]
对比例8
[0086]
本对比例与实施例6的不同在于,式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为2:1,式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩尔比为1:5,其余和实施例6相同。
[0087]
对比例9
[0088]
本对比例与实施例6的不同在于,式(ⅳ)所示化合物和硼烷四氢呋喃溶液的摩尔比为1:8,式(ⅲ)所示化合物和2-氟-6-甲基苯甲酰氯的摩尔比为2:1,其余和实施例6相同。
[0089]
按照上述实施例6-9和对比例6-9制得式(ⅱ)所示化合物,其纯度、ee%和两步收率如表3所示。
[0090]
表3
[0091] 产物纯度ee%两步收率实施例698.5%99.0%70%实施例798.3%99.2%72%实施例898.5%99.4%74%实施例998.9%99.2%75%对比例694.3%88.4%65%对比例794.3%87.8%65%对比例894.4%88.2%68%对比例994.3%87.6%66%
[0092]
上述结果表明,实施例6-9中各原料组分的比例在本发明的保护范围内,制得的产物纯度均在98.3%以上,ee%均在99%以上,证明非对应异构体的量极少,且两步收率均在70%以上。而对比例6-9中部分原料组分的比例不在本发明的保护范围内,其产物纯度低,非对应异构体的含量高且产率低。
[0093]
实施例10-12 一种avacopan中间体式(ⅰ)所示化合物的制备方法。
[0094]
基础实施例:式(ⅰ)所示化合物的制备方法,由式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺发生胺酯交换反应得到。
[0095][0096]
实施例10
[0097]
在250ml的三口瓶中加入4-甲基-3-三氟甲基苯胺(12.2g,0.07mol),和150ml二氯甲烷搅拌溶解。-5-5℃滴加2m三甲基铝的正庚烷溶液70ml,滴加时间约0.5h;滴加完毕后,20-30℃搅拌,反应0.5h;20~40℃滴加式(ⅱ)所示化合物的二氯甲烷溶液(14.5g,0.035mol溶解于75ml二氯甲烷)滴加完毕后40-50℃反应4-6h;冷却至0-10℃,滴加2m氢氧化钠溶液(100ml)淬灭反应;分液收集有机相,水相用二氯甲烷(50ml*2)萃取两次;合并有机相,分别用1m盐酸(50ml)和水(50ml)洗涤一次;减压浓缩至不滴,加入异丙醇(100ml),升温至80-90℃,搅拌0.5h至固体全部溶解,自然冷却至10-20℃,过滤干燥得到15.2g式(ⅰ)所
示化合物。
[0098]
实施例11
[0099]
本实施例与实施例10的不同在于,式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺的摩尔比为1:1,其余和实施例10相同。
[0100]
实施例12
[0101]
本实施例与实施例10的不同在于,式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺的摩尔比为1:4,其余和实施例10相同。
[0102]
对比例10
[0103]
本对比例与实施例10的不同在于,式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺的摩尔比为2:1,其余和实施例10相同。
[0104]
对比例11
[0105]
本对比例与实施例10的不同在于,式(ⅱ)所示化合物和4-甲基-3-三氟甲基苯胺的摩尔比为1:6,其余和实施例10相同。
[0106]
按照上述实施例10-12和对比例10-11制得式(ⅱ)所示化合物,其纯度、ee%和收率如表4所示。
[0107]
表4
[0108] 产物纯度ee%收率实施例1099.0%99.5%80%实施例1199.2%99.3%81%实施例1299.1%99.8%80%对比例1094.3%88.1%73%对比例1194.4%88.2%75%
[0109]
上述结果表明,实施例10-12中各原料组分的比例在本发明的保护范围内,制得的产物纯度均在99%以上,ee%均在99%以上,证明非对应异构体的量极少,且收率均在80%以上。而对比例10-11中部分原料组分的比例不在本发明的保护范围内,其产物纯度低,非对应异构体的含量高且产率低。
[0110]
实施例13-16 一种avacopan的制备方法。
[0111]
基础实施例:一种avacopan的制备方法,由式(ⅰ)所示化合物和环戊酮通过还原胺化反应制备得到,所述还原胺化反应是在钯碳的催化和氢气的参与下进行。
[0112][0113]
实施例13
[0114]
在250ml的三口瓶中加入式(ⅰ)所示化合物(15.2g,0.028mol)和150ml甲醇搅拌溶解;加入环戊酮(4.7g,0.056mol),氮气置换三次,加入10%钯碳(14.9g,0.014mol),常压通氢气24h;用氮气置换氢气,过滤除去钯碳,滤液减压浓缩至不滴;加入乙醇100ml,升温至80-90℃溶解,80-90℃下滴加水20ml,缓慢冷却至20-30℃,过滤干燥得到12.2g化合物
avacopan。
[0115]
实施例14
[0116]
本实施例与实施例13的不同在于,式(ⅰ)所示化合物和环戊酮的摩尔比为1:1,式(ⅰ)所示化合物和钯碳的摩尔比为10:1,其余和实施例13相同。
[0117]
实施例15
[0118]
本实施例与实施例13的不同在于,式(ⅰ)所示化合物和环戊酮的摩尔比为1:4,式(ⅰ)所示化合物和钯碳的摩尔比为100:1,其余和实施例13相同。
[0119]
实施例16
[0120]
本实施例与实施例13的不同在于,式(ⅰ)所示化合物和环戊酮的摩尔比为1:3,式(ⅰ)所示化合物和钯碳的摩尔比为40:1,其余和实施例13相同。
[0121]
对比例12
[0122]
本对比例与实施例13的不同在于,式(ⅰ)所示化合物和环戊酮的摩尔比为2:1,其余和实施例13相同。
[0123]
对比例13
[0124]
本对比例与实施例13的不同在于,式(ⅰ)所示化合物和钯碳的摩尔比为120:1,其余和实施例13相同。
[0125]
对比例14
[0126]
本对比例与实施例13的不同在于,式(ⅰ)所示化合物和环戊酮的摩尔比为1:6,式(ⅰ)所示化合物和钯碳的摩尔比为120:1,其余和实施例13相同。
[0127]
对比例15
[0128]
本对比例与实施例13的不同在于,式(ⅰ)所示化合物和环戊酮的摩尔比为2:1,式(ⅰ)所示化合物和钯碳的摩尔比为8:1,其余和实施例13相同。
[0129]
按照上述实施例13-15和对比例12-15制得化合物avacopan,其纯度、ee%和收率如表5所示。
[0130]
表5
[0131] 产物纯度ee%收率实施例1399.7%99.8%75%实施例1499.5%99.7%78%实施例1599.4%99.6%77%实施例1699.8%99.8%79%对比例1294.4%88.7%70%对比例1394.3%88.5%71%对比例1494.3%89.6%69%对比例1594.0%88.4%70%
[0132]
上述结果表明,实施例13-16中各原料组分的比例在本发明的保护范围内,制得的产物纯度均在99%以上,ee%均在99%以上,证明非对应异构体的量极少,且收率均在75%以上。而对比例12-15中部分原料组分的比例不在本发明的保护范围内,其产物纯度低,非对应异构体的含量高且收率低。
[0133]
最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护
范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1