一种石蜡包埋组织的DNA提取试剂盒以及提取方法与流程
一种石蜡包埋组织的dna提取试剂盒以及提取方法
技术领域
1.本发明涉及一种石蜡包埋组织的dna提取试剂盒以及提取方法,属于基因检测技术领域。
背景技术:
2.恶性胶质瘤包括间变性少突胶质细胞瘤、间变性星形细胞瘤、间变性少突星形细胞瘤及胶质母细胞瘤。恶性胶质瘤接受甲基苄肼/环己亚硝脲/长春新碱(pcv)联合化疗方案治疗中发现,1号染色体短臂(1p)杂合缺失及联合19号染色体长臂(19q)杂合缺失的间变性少突胶质细胞瘤的预后明显好于同级别的星型细胞瘤;并在随后的研究中发现1p/19q的联合缺失与化疗的敏感性相关,且1p/19q联合缺失的间变少突胶质瘤具有较长的生存期。因此1p/19q杂合缺失可以用于少突胶质细胞肿瘤的管理,在临床上作为评价诊断、治疗及预后判断的指标。替莫唑胺为第二代烷化剂,一直以来被用于恶性胶质瘤的化学治疗。替莫唑胺易通过血脑屏障且毒副作用和不良反应小。06
‑
甲基鸟嘌呤
‑
dna
‑
甲基转移酶(o6
‑
methyl guanine
‑
dna
‑
methyl transferase,mgmt)具有dna修复作用,能导致替莫唑胺耐药,即移除dna上鸟嘌呤o6位点的烷基化加合物,使损伤的鸟嘌呤恢复。mgmt基因在肿瘤细胞株沉默通常伴随着基因启动子cpg岛的甲基化,说明基因启动子甲基化介导肿瘤细胞mgmt蛋白表达。mgmt基因启动子甲基化在临床上被用于预测胶质母细胞瘤患者替莫唑胺化疗敏感性及预后情况。
3.在现有技术中,对mgmt基因启动子甲基化的检测方法主要可以分为:(1)焦磷酸测序法、(2)甲基化敏感的限制性内切酶法(ms
‑
re)、(3)荧光pcr方法、(4)高通量测序法;上述的方法中,焦磷酸测序法是“金标准”方法;而高通量测序中,目前存在着操作繁琐,引物扩增效率差,成本较高等问题。为了解决以上问题,本发明提供了一种用于检测mgmt启动子99个cpg位点甲基化的方法。
技术实现要素:
4.本发明的目的是:提供了一种对mgmt启动子上的99个cpg位点的甲基化水平进行检测的方法以及相关的试剂。
5.技术方案是:
6.一种用于扩增mgmt启动子上cpg位点的引物组,包括上游引物和下游引物,上游引物的核苷酸序列如seq id no.1
‑
4所示,下游引物的核苷酸序列如seq id no.5
‑
8所示。
7.一种mgmt启动子区甲基化检测文库的构建方法,包括以下步骤:
8.步骤1,获取样本并提取dna,对dna通过甲基化修饰转化试剂进行处理;
9.步骤2,通过引物组对步骤1中得到的核苷酸进行多重pcr扩增,获得扩增产物;
10.步骤3,对扩增产物进行末端修补、末端加碱基a、加接头后进行扩增,再经过纯化后,得到文库。
11.所述的步骤1中,样本是ffpe组织样本,提取dna是通过磁珠法获取得到。
12.所述的步骤2中,多重pcr扩增过程执行以下扩增程序:
13.92
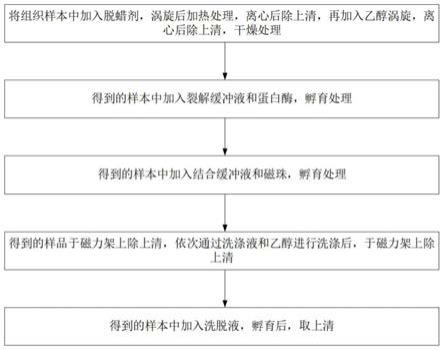
‑
97℃预变性5
‑
15min;
14.92
‑
97℃变性20
‑
40s,40
‑
65℃退火20
‑
40s,70
‑
75℃延伸0.5
‑
5min,共计30
‑
48个循环;
15.70
‑
75℃终延伸3
‑
8min。
16.所述的步骤3中,末端修补过程中的反应液中包括:dna 30
‑
70ul、磷酸化反应缓冲液5
‑
15ul、dntp 10nm、t4dna聚合酶1
‑
10ul、klenow大肠杆菌聚合酶片段0.5
‑
2ul、t4多聚核苷酸激酶1
‑
10ul、无核酸酶水将总体积补至100ul。
17.所述的步骤3中,末端加碱基a的反应液中包括:dna 20
‑
40ul、datp 1mm、10xklenow大肠杆菌聚合酶缓冲液1
‑
10ul、klenow大肠杆菌聚合酶片段1
‑
5ul、无核酸酶水将总体积补至50ul。
18.所述的步骤3中,扩增程序95
‑
99℃预变性20
‑
40秒,95
‑
99℃变性20
‑
40秒,60
‑
70℃退火20
‑
40秒,70
‑
75℃延伸20
‑
40秒,共循环3~10次,最后在70
‑
75℃延伸3
‑
6分钟。
19.所述的步骤4中,扩增过程中的反应液组成是:加上接头后的dna文库20
‑
40ul、10x高保真dna聚合酶缓冲液2
‑
6ul、dna聚合酶缓冲液0.5
‑
2ul、接头正引物1ul、接头反引物1ul、无核酸酶水将总体积补至50ul。
20.一种mgmt启动子甲基化检测方法,包括如下步骤:
21.通过上述方法获得测序文库,使用二代测序法获得发生了甲基化的cpg位点和未甲基化的cpg位点的reads数,计算出甲基化率。
22.一种试剂盒,包括有上述的引物组。
23.一种石蜡包埋组织的dna提取试剂盒,包括:
24.脱蜡剂,用于对样本进行脱蜡处理;
25.裂解缓冲液和蛋白酶,用于对样本进行消化处理;
26.结合缓冲液和磁珠、用于对消化产物中的dna进行结合;
27.洗涤液和乙醇,用于对结合了dna的磁珠进行洗涤;
28.洗脱液,用于将磁珠上结合的dna进行洗脱。
29.所述的脱蜡剂是二甲苯。
30.所述的裂解缓冲液中包含:5
‑
50mmol/l的tris
‑
hcl;1
‑
5mmol/l的nacl;5
‑
20mmol/l的edta;8
‑
50mmol/l的dtt;3
‑
5mol/l的异硫氰酸胍;0.5%
‑
2%的sds;1%
‑
5%的tritonx
‑
100。
31.所述的蛋白酶是蛋白酶k。
32.所述的组合缓冲液中包含:100
‑
200mmol/l的tris
‑
hcl;1
‑
5mmol/l的nacl;1%
‑
5%的tritonx
‑
100;2
‑
4mol/l的异硫氰酸胍;20
‑
40%的异丙醇。
33.所述的洗涤液包括第一洗涤液和第二洗涤液,第一洗涤液中包含:100
‑
200mmol/l的tris
‑
hcl;1
‑
5mmol/l的nacl;40
‑
60%的乙醇;2
‑
4mol/l的异硫氰酸胍;第二洗涤液中包含:25
‑
75mol/l的tris
‑
hcl;75
‑
85%的乙醇。
34.所述的乙醇是无水乙醇。
35.所述的洗脱液无核酸酶水。
36.一种石蜡包埋组织的dna提取方法,包括如下步骤:
37.步骤1,将组织样本中加入脱蜡剂,涡旋后加热处理,离心后除上清,再加入乙醇涡旋,离心后除上清,干燥处理;
38.步骤2,步骤1中得到的样本中加入裂解缓冲液和蛋白酶,孵育处理;
39.步骤3,步骤2中得到的样本中加入结合缓冲液和磁珠,孵育处理;
40.步骤4,步骤4中得到的样品于磁力架上除上清,依次通过洗涤液和乙醇进行洗涤后,于磁力架上除上清;
41.步骤5,步骤4中得到的样本中加入洗脱液,孵育后,取上清。
42.所述的步骤1中,涡旋时间5
‑
20s,离心力10000
‑
20000g,单次离心时间1
‑
15min;加热处理中是在35
‑
60℃处理5
‑
20min。
43.所述的步骤2中,孵育处理过程是45
‑
65℃、500
‑
1500rpm转速下处理1
‑
2h,再升温至85
‑
95℃,在200
‑
500rpm转速下处理5
‑
15min。
44.所述的步骤3中,孵育处理是室温下处理1
‑
30min。
45.所述的步骤4中,洗涤过程中依次采用第一洗涤液、第二洗涤液和乙醇洗涤,每次洗涤中涡旋1
‑
30s,并孵育1
‑
5min。
46.所述的步骤5中,孵育1
‑
10min。
47.有益效果
48.本专利的方法可以有效地对mgmt启动子上的99个cpg位点的甲基化水平进行检测,具有检测准确性高、覆盖范围广的优点。
附图说明
49.图1是本专利的流程图;
50.图2是dna样本提取流程;
51.图3是实施例1的各个位点的测序深度图;
52.图4是实施例1的甲基化水平检测结果;
53.图5是对照例1的各个位点的测序深度图;
54.图6是对照例1的甲基化水平检测结果;
具体实施方式
55.样本来源于脑胶质瘤患者ffpe组织,甲基化阴性对照样品为健康人ffpe组织,甲基化阳性对照样品为细胞系sw620。
56.首先从样本中对dna进行提取,从样本中提取基因组dna。组织样本提取试剂盒以及提取步骤如下:
57.1)利用脱蜡剂将ffpe样本进行脱蜡,以便获得脱蜡的样本;进一步可以包括利用乙醇洗涤去除脱蜡剂,能够有效地去除脱蜡剂,易于后续处理。脱蜡剂可以采用二甲苯。乙醇优选无水乙醇,
58.2)利用裂解缓冲液和蛋白酶k对脱蜡的ffpe样本或新鲜组织和细胞进行消化处理,以便获得消化产物,消化产物中含有释放的dna。裂解缓冲液的组成包括:5
‑
50mmol/l的tris
‑
hcl;1
‑
5mmol/l的nacl;5
‑
20mmol/l的edta;8
‑
50mmol/l的dtt;3
‑
5mol/l的异硫氰酸胍;0.5%
‑
2%的sds;1%
‑
5%的tritonx
‑
100。其中蛋白酶k使用量为0.5
‑
2.5mg。优选地,裂
解液组成:25mmol/l的tris
‑
hcl;2.5mmol/l的nacl;10mmol/l的edta;25mmol/l的dtt;4mol/l的异硫氰酸胍;1%的sds;5%的tritonx
‑
100,以及蛋白酶k1.5mg。
59.3)在消化产物中加入结合缓冲液及磁珠,得到磁珠dna结合产物,以便磁珠于dna结合。其中结合缓冲液的组成包括:100
‑
200mmol/l的tris
‑
hcl;1
‑
5mmol/l的nacl;1%
‑
5%的tritonx
‑
100;2
‑
4mol/l的异硫氰酸胍;20
‑
40%的异丙醇。根据本发明的具体示例,该结合液优选:150mmol/l的tris
‑
hcl;2.5mmol/l的nacl;2.5%的tritonx
‑
100;3mol/l的异硫氰酸胍;30%的异丙醇。
60.4)使用洗涤液1、洗涤液2及无水乙醇分别对磁珠dna结合产物进行清洗,以便回收纯化dna。其中洗涤液1的组成:100
‑
200mmol/l的tris
‑
hcl;1
‑
5mmol/l的nacl;40
‑
60%的乙醇;2
‑
4mol/l的异硫氰酸胍。根据本发明的具体示例,该结合液优选:150mmol/l的tris
‑
hcl;2.5mmol/l的nacl;60%乙醇;3mol/l的异硫氰酸胍。其中洗涤液2的组成:25
‑
75mol/l的tris
‑
hcl;75
‑
85%的乙醇。根据本发明的具体示例,该结合液优选:50mmol/l的tris
‑
hcl;80%乙醇。
61.5)使用无核酸酶水从磁珠上将dna进行洗脱,以便得到纯化的dna。
62.操作步骤如下:
63.1)脑胶质瘤患者ffpe组织和健康人ffpe组织样本脱蜡处理。
①
使用切片机从石蜡包埋组织组织块中切割切片,收集在1.5ml ep管中。
②
向切片中加入1ml二甲苯涡旋15s,再将样品放置在50℃金属浴上加热10min,18000g速度离心5min,去除上清。
③
加入1ml 100%乙醇涡旋15s,18000g速度离心5min,去除上清。重复步骤
③
。
④
使用真空干燥仪45℃真空干燥5
‑
10min,取出样本。
64.2)细胞系sw620离心处理。
①
将细胞使用pbs重悬,收集在1.5ml ep管中。
②
300g速度离心5min,去除上清。
65.3)向已处理ffpe和细胞样本中加入380μl裂解缓冲液和20μl蛋白酶k,涡旋振荡5s,56℃800rpm孵育1
‑
2h,具体情况视组织完全消化时间而定。
66.4)转移至温度已升至90℃350rpm孵育10min。
67.5)短暂离心,待样本恢复室温后加入380μl结合缓冲液和20μl磁珠,涡旋混匀30秒,室温旋转孵育10min。
68.6)置于磁力架上静置3min,静置1.5min时上下颠倒一次样本管,去除上清。
69.7)从磁力架上取下,加入500μl洗涤缓冲液1,涡旋混匀10s,室温孵育2min。
70.8)置于磁力架上静置3min,静置1.5min时上下颠倒一次样本管,去除上清。
71.9))从磁力架上取下,加入500μl洗涤缓冲液2,涡旋混匀10s,室温孵育2min。
72.10)置于磁力架上静置3min,静置1.5min时上下颠倒一次样本管,去除上清。
73.11)从磁力架上取下,加入500μl100%乙醇,涡旋混匀10s,室温孵育2min。
74.12)置于磁力架上静置3min,静置1.5min时上下颠倒一次样本管,去除上清。
75.13)待磁珠干燥后将样本管从磁力架上取下,沿着管壁向磁珠上方加入100μl无核酸酶水,涡旋混匀,使磁珠从管壁上分离,完全混匀于无酶水中,室温孵育5min。
76.14)将样本管置于磁力架上静置2min,静置完成后,小心转移上清液至新的1.5ml离心管中。
77.在获得了样本dna后,进行亚硫酸氢盐转化与回收,将提取的基因组dna样本中添
加内部对照,然后使用ez dna methylation
‑
gold kit(zymo research公司)试剂盒将含有内部对照的dna样本进行转化,具体提取步骤参见试剂盒操作说明书;
78.以转化后的基因组dna为模板,使用引物组合进行mgmt启动子区域甲基化特异性多重pcr扩增,扩增mgmt启动子区域cpg岛序列组合包括4条上游引物,4条下游引物,具体序列如下:
79.本专利中的引物设计构思是:4对引物扩增区域覆盖mgmt启动子99个cpg位点,位于启动子5’调控区域的
‑
552bp~+289bp处(位点的定义是:相对于转录起始位点(transcriptional start site,tss)的距离,上游为负,下游为正)
‑
552bp~+289bp,引物长度为20~25bp,引物自身需避免覆盖甲基化cpg点,引物对间注意退火温度差距≤5℃,且避免引物对间形成引物二聚体。
80.表1引物设计
[0081][0082]
表2多重pcr扩增程序
[0083][0084][0085]
3)将获得的多重pcr扩增产物进行磁珠纯化;
[0086]
文库构建是为测序片段添加接头的过程。扩增后的dna片段两端需要加上接头才能进行上机测序。文库构建主要包括以下几个步骤:
[0087]
1)dna末端修补。将dna片段进行末端修复可以利用klenow片段、t4dna聚合酶和t4多核苷酸激酶进行。利用t4聚合酶及klenow大肠杆菌聚合酶片断,对于cdna/dna5’突出粘末端补平以及3’突出粘末端打平,产生平末端,用于后续的平端连接。根据本发明的实施例,还可以进一步包括对具有粘性末端a的dna片段进行纯化的步骤,由此能够方便地进行后续处理。反应在pcr扩增仪中进行,20℃
‑
30分钟。反应体系如下表:
[0088]
表3 dna末端修补反应液组成
[0089][0090]
2)在cdna/dna样本3’末端加上碱基a。在经过末端修复的dna片段的3’末端添加碱基a,以便获得具有粘性末端a的dna片段。根据本发明的实施例,还可以进一步包括对具有粘性末端a的dna片段进行纯化的步骤,由此能够方便地进行后续处理。
[0091]
反应在pcr扩增仪中进行,37℃,30分钟。反应体系如下表:
[0092]
表4 dna末端加a反应液组成
[0093][0094]
3)在dna两端加上接头。利用t4连接酶将接头连接到dna片段两端,用于后续文库扩增。
[0095]
表5加接头反应液组成
[0096][0097]
4)dna文库扩增。
[0098]
聚合酶链反应(pcr),在pcr扩增仪中进行,反应程序:98℃预变性30秒,98℃变性30秒,65℃退火30秒,72℃延伸30秒,共循环4~6次。最后在72℃延伸5分钟。反应体系如下表:
[0099]
表6文库扩增反应液组成
[0100][0101]
5)文库纯化。将扩增的文库dna片段利用磁珠纯化出来,用于测序上机。使用
beckman coulter ampure beads试剂盒(货号:a63880)进行磁珠法纯化。
[0102]
5、甲基化检测方法
[0103]
使用illumina测序平台对甲基化文库进行测序。测序完成下机后,使用bcl2fastq生成fastq原始序列,再使用trimmomatic对原始数据进行质控,去除接头和低质量的碱基。得到的cleandata使用bismark进行基因组(hg19)的比对。比对后得到发生了甲基化的cpg位点,再根据获得的位点确定出在每个cpg位点甲基化的reads数和在这个位点区域上未甲基化的reads数,获得mgmt启动子区域甲基化率。
[0104]
甲基化百分比=甲基化测序数/(甲基化测序数+非甲基化测序数)
×
100%
[0105]
mgmt启动子区域测序深度如图3所示。对临床样本的甲基化测序结果如图4所示,本发明可以对mgmt启动子区域99个cpg位点进行甲基化水平检测,计算样本平均甲基化水平,另外利用焦磷酸测序验证其中8个cpg位点甲基化水平,本发明与焦磷酸测序结果相似。
[0106]
表6甲基化水平测试结果
[0107][0108][0109]
对两例临床样本的引物组合优化结果如上所示。
[0110]
对照例1
[0111]
用于扩增mgmt启动子区域cpg岛序列的甲基化位点检测的引物序列组合,包括3对引物组合,分别为:上游引物f1、下游引物r1,上游引物f2、下游引物r2,上游引物f3、下游引物r4,具体序列如下:
[0112]
表7引物设计
[0113][0114]
其余条件如实施例1。
[0115]
mgmt启动子区域测序深度如图5所示。mgmt启动子区域中每一个cpg岛序列甲基化水平,计算样本平均甲基化水平,如图6所示。
[0116]
表8甲基化测试结果
[0117][0118]
优化后的引物组合能够明显提高ngs测序总体测序深度,并且与焦磷酸法中获得的数据偏差更小。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1