一种本芴醇粗品的精制工艺的制作方法
1.本发明属于本芴醇合成技术领域,具体涉及本芴醇粗品的精制工艺技术领域。
背景技术:
2.本芴醇是军事医学科学院邓蓉仙团队首创的抗疟药物。本芴醇对间日疟有性体和无性体有明显的杀灭作用,对间日疟有良好的防止作用,对恶性疟无性体也有杀灭作用。但本芴醇起效缓慢,中国军事医学科学院研究人员将其与抗疟起效快的蒿甲醚配伍,二者抗疟作用可以互补。
3.邓蓉仙团队在专利申请文件cn 1029680c中公开了本芴醇五步法合成路线,即将芴在冰醋酸中氯化得到2,7-二氯芴,再以三氯化铝催化酰化得到2,7
‑ꢀ
二氯-4-氯乙酰基-芴,然后以硼氢化钾还原和强氧化钾环化得到2,7-二氯-4
‑ꢀ
环氧乙烷基-芴,该化合物和二正丁胺在高温下反应得到α-(二正丁胺基)-2, 7-二氯-4-芴甲醇,最后该中间体在乙醇中与对氯苯甲醛缩合得到本芴醇粗品,本芴醇粗品再经精制得到质量合格的本芴醇。
4.在其后众多本芴醇和其中间体合成报道中均沿用了该合成路线。申请人生产的本芴醇粗品也同样沿用了邓蓉仙团队的五步法合成路线,在此基础上做了改进,主要步骤有:以α-(二正丁胺基)-2,7-二氯-4-芴甲醇和对氯苯甲醛为原料,甲醇为溶剂,甲醇钠为碱性催化剂,加热回流反应,再降温离心得到本芴醇粗品。本芴醇粗品纯度为97%-98%,单杂为0.5%-1.3%。本芴醇粗品中含有的杂质主要有α-(二正丁胺基)-2,7-二氯-4-芴甲醇、对氯苯甲醛、对氯苯甲酸、甲醇、氢氧化钠(微量)等。本芴醇五步法合成路线由于减少了对各个中间体提纯的步骤,导致最终产物本芴醇粗品的杂质含量增加,对本芴醇粗品精制工艺提出了更高的要求。
5.专利申请文件cn 101747210 a公开了制备α-(二正丁氨基甲基)-2,7
‑ꢀ
二氯-4-芴甲醇及其盐酸盐的方法,其中采用乙酸乙酯来洗涤中间产物α-(二正丁氨基甲基)-2,7-二氯-4-芴甲醇,但由于处理对象并非本芴醇粗品,因此不具有参考意义。
6.而专利申请文件cn 103319356 a公开了抗疟疾原料本芴醇一步法绿色合成工艺,也是通过α-(二正丁胺基)-2,7-二氯-4-芴甲醇和对氯苯甲醛缩合制得本芴醇粗品,再加入丙酮进行重结晶。但丙酮安全风险高,采购手续复杂,不易获得,因此不利于工业化大规模生产;其次,重结晶后得到的本芴醇产品纯度为99%,还有一定提高的空间。因此,仍需要对现有技术中本芴醇粗品的精制工艺作进一步改进。
技术实现要素:
7.针对现有技术中存在的技术问题,本发明提供了一种本芴醇粗品的精制工艺。
8.本发明采用的技术方案如下:
9.1.一种本芴醇粗品的精制工艺,其特征在于,包括以下步骤:
10.s1、使用有机溶剂溶解本芴醇粗品;
11.有机溶剂包括乙酸乙酯、正丁醇、甲醇、二氯甲烷中的一种或多种的组合;
12.s2、待本芴醇粗品完全溶解后加水,充分搅拌,静置分层;
13.s3、将有机相趁热过滤;
14.s4、滤液浓缩,然后边搅拌边降温结晶,抽滤得到本芴醇精制湿品;
15.s5、烘干、粉碎得到本芴醇成品。
16.采用以上技术方案,关键步骤在于s1和s2,本发明步骤s1中选用的几种有机溶剂对本芴醇以及其中的杂质,如中间体α-(二正丁胺基)-2,7-二氯-4
‑ꢀ
芴甲醇、对氯苯甲醛、甲醇等都有不错的溶解性。步骤s2加水搅拌静置分层时,水溶性更强的对氯苯甲醛、甲醇等转移至水相,部分中间体α-(二正丁胺基)
ꢀ‑
2,7-二氯-4-芴甲醇也随甲醇转移至水相中。由于本芴醇的水溶性不强,所以本芴醇产品大部分溶解于有机相中。本发明中有机溶剂和水的加入顺序不可调换,否则本芴醇和可溶性杂质无法充分分离。在经过静置分层后,分离出有机相,再将有机相过滤,将不溶于有机相的固体物质过滤出来。步骤s2和s3使得s4中滤液里杂质大量减少。
17.需要说明的是,本发明和背景技术中专利申请文件cn 103319356 a涉及的本芴醇精制工艺工作原理不同。专利申请文件cn 103319356 a是利用丙酮对本芴醇及其杂质的良好溶解性进行重结晶进而达到精制的目的,而本技术则是利用有机溶剂和水之间极性的差异进行萃取,从而达到精制的目的。
18.本发明的精制对象本芴醇粗品,是申请人沿用了邓蓉仙团队的五步法合成路线合成的,在此基础上做了改进合成的,主要步骤有:以α-(二正丁胺基)
ꢀ‑
2,7-二氯-4-芴甲醇和对氯苯甲醛为原料,甲醇为溶剂,甲醇钠为碱性催化剂,加热回流反应,再降温离心得到本芴醇粗品。本芴醇粗品的纯度为97%-98%,单杂为0.5%-1.3%,本芴醇粗品中含有的杂质主要有α-(二正丁胺基)-2,7
‑ꢀ
二氯-4-芴甲醇、对氯苯甲醛、对氯苯甲酸、甲醇、氢氧化钠(微量)等。需要说明的是,本发明的本芴醇粗品实际上是湿品,其中残余有约18%的甲醇溶剂。本领域技术人员应当理解,采用其他方法合成的本芴醇粗品可能含有与本发明中不同的杂质,因此其对应采用的精制方法也不会对本发明产生启示。
19.关于有机溶剂的选用问题,本发明不仅需要考虑对本芴醇及其杂质的溶解性,还需要考虑有机溶剂的极性问题,优选弱极性溶剂。常用溶剂的极性顺序参考如下:水》甲酰胺》三氟乙酸》dmso》乙腈》dmf》六甲基磷酰胺》甲醇》乙醇》乙酸》异丙醇》吡啶》四甲基乙二胺》丙酮》三乙胺》正丁醇》二氧六环》四氢呋喃》甲酸甲酯》三丁胺》甲乙酮》乙酸乙酯》氯仿》三辛胺》碳酸二甲酯》乙醚》异丙醚》 正丁醚》三氯乙烯》二苯醚》二氯甲烷》二氯乙烷》苯》甲苯》四氯化碳》二硫化碳》 环己烷》己烷》煤油(石油醚)。
20.本发明认为氯仿也能很好的满足以上所述的有机溶剂选用标准,但由于氯仿和丙酮一样原料不易得,因此本发明没有选择氯仿作为有机溶剂。
21.采用步骤s1中所述的有机溶剂精制得到的本芴醇产品纯度可以达到99.6%以上,单杂控制在0.22%以内。考虑到进一步提高本芴醇产品的收率、纯度,同时降低单杂的含量,需要优化步骤s1中有机溶剂和本芴醇粗品的比例。列举以下几种方案并分情况进行讨论:
22.方案i:有机溶剂为乙酸乙酯,且乙酸乙酯与本芴醇粗品质量比优选为 (6-7):1,更优选为6:1。
23.方案ii:有机溶剂为正丁醇,且正丁醇与本芴醇粗品质量比优选为(8-9): 1,更优
选为8:1。
24.方案iii:有机溶剂为乙酸乙酯和正丁醇的混合溶剂,且混合溶剂与本芴醇粗品质量比优选为(8-9):1,更优选为8:1,乙酸乙酯和正丁醇质量比优选为(4-7):1,更优选为(6-7):1。
25.方案iv:有机溶剂为乙酸乙酯和甲醇的混合溶剂,且混合溶剂与本芴醇粗品质量比优选为(8-9):1,更优选为8:1,乙酸乙酯和甲醇质量比优选为(3-7): 1,更优选为(5-6):1。
26.以上四种优选方案得到的精制本芴醇产品纯度能够达到99.7%以上,单杂控制在0.1%以内,以上所述更优选方案得到的精制本芴醇产品纯度能够达到 99.8%以上,单杂控制在0.08%以内。
27.进一步,步骤s4具体为:常压浓缩至本芴醇在溶液中析晶,然后80-90r/min 低速搅拌,同时控制降温速度以10-5℃/h梯度降温,最后降温至25℃, 50-60r/min低速搅拌3-3.5h,抽滤得到本芴醇精制湿品。更进一步,常压浓缩至有机溶剂与本芴醇粗品的质量比为(2.5-3.0):1,此时本芴醇在溶液中析晶。
28.步骤s4中浓缩结晶阶段,采用低速搅拌的方式,是为了保证结晶体晶形完整,晶体大;同时控制降温速度,是为了保证结晶过程不形成晶包杂的情况。
29.进一步,步骤s5中,烘干过程具体分为以下两个阶段:s5-1、先在40℃、-0.10mpa的低温高真空条件下烘干2h,从而将精制湿品中的有机溶剂抽出,再于80℃、-0.08mpa的高温低真空条件下烘干6h,将残余水分烘干。
30.相较于现有技术,本发明的有益效果是:
31.(1)本发明采用的有机溶剂均原料来源广泛,采购手续简单,容易获取,方便生产;
32.(2)本发明利用极性不同的有机溶剂和水两相体系对本芴醇粗品进行萃取提纯,提高了精制本芴醇产品的纯度;
33.(3)本发明采用先加有机溶剂后加水的方式,使得本芴醇和可溶性杂质得到充分分离,调换添加顺序后则分离效果较差,得到的本芴醇产品纯度很低;
34.(4)本发明在萃取后进行不溶性杂质的过滤,大大降低了滤液中可溶性和不可溶性杂质的含量;
35.(5)本发明采用边搅拌边冷却结晶的方式,获得的结晶体晶形完整,晶体大;
36.(6)本发明精制得到的本芴醇产品的纯度为99.6%以上,单杂含量低于 0.22%。
具体实施方式
37.以下示出实施例以及比较例更详细地说明本发明。本发明不限于以下的实施例。
38.以下实施例和对比例的精制对象本芴醇粗品,是沿用了邓蓉仙团队的五步法合成路线合成的,在此基础上做了改进,主要步骤有:以α-(二正丁胺基)
ꢀ‑
2,7-二氯-4-芴甲醇和对氯苯甲醛为原料,甲醇为溶剂,甲醇钠为碱性催化剂,加热回流反应,再降温离心得到本芴醇粗品。本芴醇粗品纯度为97.4%,单杂为0.9%,残余溶剂甲醇18%。本芴醇粗品中含有的杂质主要有α-(二正丁胺基)
ꢀ‑
2,7-二氯-4-芴甲醇、对氯苯甲醛、对氯苯甲酸、甲醇、氢氧化钠(微量)等。
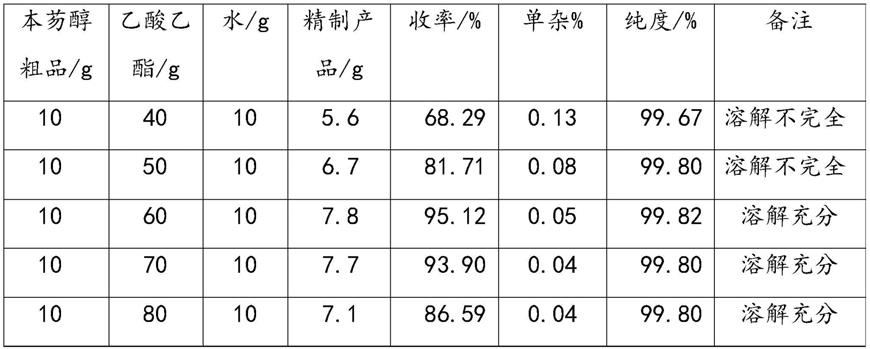
39.实施例1[方案i:乙酸乙酯+水]
[0040]
s1、使用有机溶剂乙酸乙酯溶解本芴醇粗品;
[0041]
s2、待本芴醇粗品完全溶解后加水,充分搅拌,静置分层,分离出有机相;
[0042]
s3、将有机相趁热经过两级布袋过滤器过滤;
[0043]
s4、滤液常压浓缩至本芴醇在溶液中析晶,此时有机溶剂与本芴醇粗品的质量比约为(2.5-3.0):1;然后85r/min低速搅拌,同时控制降温速度以8℃ /h梯度降温,最后降温至25℃,55r/min低速搅拌3h;抽滤得到本芴醇精制湿品;
[0044]
s5、烘干、粉碎得到本芴醇成品,其中烘干过程分以下两个阶段进行:s5-1、先在40℃、-0.10mpa条件下烘干2h,再于80℃、-0.08mpa条件下烘干6h。
[0045]
实施例1采用的乙酸乙酯和水的添加量以及相应的精制本芴醇产品的质量、收率、纯度、单杂情况如表1所示。
[0046]
表1.
[0047][0048]
注:由于本芴醇粗品是未进行烘干处理的湿品,含有残余甲醇18%,因此,本芴醇粗品中实际含本芴醇干重为10g
×
(1-18%)=8.2g,收率=精制产品质量 /8.2g
×
100%。
[0049]
由表1可知,当乙酸乙酯与本芴醇粗品比例在(6-7):1时,精制产品的收率较高,达到93.90%以上,纯度为99.8%以上,单杂在0.05%以下。
[0050]
当当乙酸乙酯与本芴醇粗品比例在6:1时,精制产品的收率最高,达到 95.12%以上,纯度为99.82%以上,单杂在0.05%以下。
[0051]
实施例2[方案ii:正丁醇+水]
[0052]
除了有机溶剂采用正丁醇,其余精制步骤与实施例1相同。实施例2采用的正丁醇和水的添加量以及相应的精制本芴醇产品的质量、收率、纯度、单杂情况如表2所示。
[0053]
表2.
[0054][0055]
由表2可知,当正丁醇与本芴醇粗品比例在(8-9):1时,精制产品的收率较高,达到了79.26%以上,纯度达到99.8%以上,单杂为0.09%以下。
[0056]
当正丁醇与本芴醇粗品比例在8:1时,精制产品的收率最高,达到了 86.59%,纯度达到99.8%,单杂为0.08%。
[0057]
实施例3[方案iii:乙酸乙酯+正丁醇+水]
[0058]
除了有机溶剂采用乙酸乙酯和正丁醇的混合溶剂,其余精制步骤与实施例1相同。实施例3采用的乙酸乙酯、正丁醇和水的添加量以及相应的精制本芴醇产品的质量、收率、纯度、单杂情况如表3所示。
[0059]
表3.
[0060][0061]
由表3可知,当混合溶剂总量8倍于本芴醇粗品,且乙酸乙酯:正丁醇比例为(4-7):1时,精制产品的收率较高,达到81.71%以上,纯度达到99.77%以上,单杂控制在0.08%以下。
[0062]
当混合溶剂总量8倍于本芴醇粗品,且乙酸乙酯:正丁醇比例为(6-7): 1时,精制产品的收率更高,达到86.59%以上,纯度达到99.77%以上,单杂控制在0.05%以下。
[0063]
当混合溶剂总量8倍于本芴醇粗品,且乙酸乙酯:正丁醇比例为6:1时,精制产品的收率最高,达到89.02%以上,纯度达到99.80%以上,单杂控制在 0.04%以下。
[0064]
实施例4[方案iv:乙酸乙酯+甲醇+水]
[0065]
除了有机溶剂采用乙酸乙酯和甲醇的混合溶剂,其余精制步骤与实施例1 相同。实施例4采用的乙酸乙酯、甲醇和水的添加量以及相应的精制本芴醇产品的质量、收率、纯度、单杂情况如表4所示。
[0066]
表4.
[0067][0068]
由表4可知,当混合溶剂总量8倍于本芴醇粗品时,乙酸乙酯:甲醇比例为(3-7):1时,精制产品收率较高,达到91.46%以上,纯度达到99.70%以上,单杂控制在0.07%以下。
[0069]
当混合溶剂总量8倍于本芴醇粗品时,乙酸乙酯:甲醇比例为(5-6):1 时,精制产品收率更高,达到93.90%以上,纯度达到了99.90%以上,单杂控制在0.04%以下。
[0070]
当混合溶剂总量8倍于本芴醇粗品时,乙酸乙酯:甲醇比例为5:1时,精制产品收率最高,达到95.12%以上,纯度达到了99.91%以上,单杂控制在0.04%以下。
[0071]
对比例1[丙酮+水]
[0072]
除了有机溶剂采用丙酮,其余精制步骤与实施例1相同。对比例1采用的丙酮和水的添加量以及相应的精制本芴醇产品的质量、收率、纯度、单杂情况如表5所示。
[0073]
表5.
[0074][0075]
通过实施例1-4和对比例1的比较发现,实施例1和4的收率、纯度以及单杂均优于对比例1的,而实施例2和3的收率低于对比例1的,纯度以及单杂与对比例1的基本相当。但本发明首要考虑的因素是精制工艺采用的溶剂原料的易得性,因此本发明不采用对比例1的方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1