通过微通道反应制备醋酸阿比特龙中间体的方法与流程
1.本发明属于化学制药领域,更具体地,本发明涉及通过微通道反应制备醋酸阿比特龙中间体、特别是17-碘-雄甾-5,16-二烯-3β-醇中间体的方法。
背景技术:
2.醋酸阿比特龙是一种口服cyp17酶不可逆抑制剂,由ceotocor ortho公司开发,用于与泼尼松联用治疗去势抵抗性前列腺癌,于2011年4月28日经美国食品药品监督管理局(fda)批准上市。与目前的常规治疗方法相比,醋酸阿比特龙疗效更好,副作用更低。
3.wo1995009178a1公开了一种制备醋酸阿比特龙的方法,合成路线如下:
[0004][0005]
该路线以醋酸去氢表雄酮为起始原料,首先和水合肼发生缩合反应,得到去氢表雄酮-17-腙中间体,经碘代反应,然后在钯催化剂存在下与二乙基-(3-吡啶基)硼烷进行偶联反应得到阿比特龙,最后再与醋酸酐酯化得到醋酸阿比特龙。
[0006]
如上述工艺路线,中间体3(17-碘-雄甾-5,16-二烯-3β-醇)的制备属于碘取代反应,文献tetrahedron letters,vol.24,no.l5,pp 1605-1608,1983公开了一种改进的制备乙烯基碘代物的方法,其中提到的改进方法包括三个方面(1)反应严格控制水分(2)使用四甲基胍作碱(3)将底物加入到碘溶液中;直接以化合物1为底物,采用四甲基胍为碱,制备得到目标碘化物收率为75%,另外会生成16%的二碘化物。
[0007][0008]
专利wo1995009178a1提供了一种技术方案,该方案使用四氢呋喃乙醚混合溶液为溶剂,加入碘,搅拌溶解降温,加入四甲基胍,搅拌。将化合物1的四氢呋喃溶液缓慢(2h)滴加入上述碘溶液中,加毕,控温0℃反应1h。后处理滤除盐后,真空浓缩成油状物,该油状物
继续在油浴中加热4h(使17,17-二碘副产物转化为产品)后,溶解杂乙醚中,乙醚溶液经稀盐酸、水洗涤后分液,干燥,浓缩结晶得到粗品,粗品经正己烷洗涤干燥后再加个乙醇水重结晶,得到目标产品,收率83.0%。该工艺在产业化中还有两个问题需要解决,第一是溶剂的用量太大,按照文献用量,化合物1投料90.74g,反应中需要用到四氢呋喃6.25l和乙醚2.0l作溶剂,后处理还要用到33倍体积的乙醚,固液比达到1:120,这种固液比无法用于大规模生产,且乙醚具有低沸点和麻醉作用,大量使用具有安全风险。由于化合物1在四氢呋喃和乙醚中的溶解度很小,改善这个问题难度很大,或者考虑改变溶剂使用方法。第二是反应后处理复杂,需要使用酸洗、水洗、盐洗及正己烷洗涤,不仅产生大量废水废液,操作工序的复杂直接导致生产效率的低下和成本的上升。
[0009][0010]
从现有技术的疑难来看,解决由化合物2到化合物3的生产问题,对于醋酸阿比特龙的产业化生产具有重要意义。
技术实现要素:
[0011]
为了克服上述问题,本发明人提供一种解决方案,即利用微通道反应制备醋酸阿比特龙中间体。
[0012]
本发明提供一种通过微通道反应制备醋酸阿比特龙中间体17-碘-雄甾-5,16-二烯-3β-醇的方法,该方法包括如下步骤:
[0013]
(1)将碘、四甲基胍溶于溶剂中,配制成碘和四甲基胍各自的浓度分别为0.2~3.0kg/l的溶液r;
[0014]
(2)将化合物3β-羟基-雄甾-5-烯-17-腙溶于溶剂中,配制成浓度为0.1~2.0kg/l的浆料s;
[0015]
(3)通过加料泵,将溶液r和浆料s按一定的动态物料配比引入到微通道反应器,进行连续反应;
[0016]
(4)将从微通道反应器中流出的反应液进行固-液分离,分离出的液体加入洗涤溶液进行洗涤,然后进行液-液分离,分出洗涤液,并将余下的有机相蒸馏浓缩,以回收溶剂;及
[0017]
(5)向得自步骤(4)的浓缩剩余物中加入混合溶剂并加热转化,得到中间体17-碘-雄甾-5,16-二烯-3β-醇。
[0018]
在根据本发明的上述制备方法的一个实施方案中,步骤(1)和(2)中所述的溶剂为选自甲基叔丁基甲醚、四氢呋喃中的一种或多种。
[0019]
在根据本发明的上述制备方法的另一个实施方案中,步骤(3)所述按一定的动态物料配比进料是指化合物3β-羟基-雄甾-5-烯-17-腙:碘:四甲基胍的当量配比为1.0eq:2.0~3.0eq:3.0~5.0eq。
[0020]
在根据本发明的上述制备方法的另一个实施方案中,步骤(5)中所述混合溶剂为乙醇和水的混合溶剂,乙醇和水的比例为1:1~10:1(重量比),且该混合溶剂的用量为化合物3β-羟基-雄甾-5-烯-17-腙的5~12倍(重量比)。
[0021]
在根据本发明的上述制备方法的另一个实施方案中,所述加热转化的温度为70℃至混合溶剂回流温度,加热转化时间为1.0h~2.0h。
[0022]
在根据本发明的上述制备方法的另一个实施方案中,步骤(5)还包括在加热转化之后对产物进行冷却结晶、离心过滤和干燥。
[0023]
通过一定比例的碘和四甲基胍溶液和化合物2的匀速定量微通道反应,除可以解决现有技术中存在的前述问题,实现一定转化率下的反应控制之外,发明人经深入研究发现,利用本发明的微通道方法生产醋酸阿比特龙关键中间体3还具有如下优点:
[0024]
(i)由于反应本身并没有明显放热(能量以氮气的方式释放),微通道反应也不会出现能量累计,因此可以采用常温反应,避免低温制冷需要的额外能源消耗。
[0025]
(ii)微通道反应器可改善釜式反应器中反应大量放出氮气的不足,在釜式反应器中,快速反应会导致氮气剧烈放出,从而可能引发冲料,而在微通道反应器中,由于反应流比较平缓,氮气的产生并不明显,通过反应流速控制可以使反应与氮气释放进行平稳控制。
[0026]
(iii)可实现副产物的回收,避免高盐废液的产生,提高经济效益和保护环境。微通道反应后直接实现副产物四甲基胍氢碘酸盐的绝大部分分离收集反应液中极少残留,因此洗涤用水将大幅减少。
[0027]
(iv)实现了溶剂的可回收利用,避免了危险试剂(乙醚)的使用。本发明专利使用甲基叔丁基甲醚代替乙醚,并大幅降低了四氢呋喃的用量,后处理淬灭后直接蒸馏,回收大部分溶剂,节约了生产成本。甲基叔丁基甲醚、四氢呋喃常压蒸馏回收率高,可反复套用。
[0028]
(v)改进了杂质转化的工艺,将纯化和杂质转化合并进行,提高了生产效率。实验研究发现,实现二碘代副产物向目标产物的转化,并不只有在油浴中加热一个方法,使用乙醇水的体系加热也可以达到二碘代副产物向目标产物转化的效果,而且转化速率更快,1h内即可完成。
[0029]
(vi)设备要求小,可实现连续化生产,大大提高了生产效率。对于醋酸阿比特龙这种规格较大(250mg/片)的药物而言,如果采用传统釜式反应,要达到一定量的生产规模对溶剂试剂和设备的要求是非常大的,而使用微通道反应系统,可以完全避免大型反应釜的使用,连续生产和集中处理协调进行,大大提高了生产效率。
[0030]
(vii)反应可控性强,可以通过配置不同浓度溶液,调节不同进料速率,实现反应的控制,操作方便。
附图说明
[0031]
图1是根据本发明的方法制备的醋酸阿比特龙关键中间体化合物3的核磁共振氢谱(h nmr);
[0032]
图2是根据本发明的方法制备的醋酸阿比特龙关键中间体化合物3的化学纯度谱图;及
[0033]
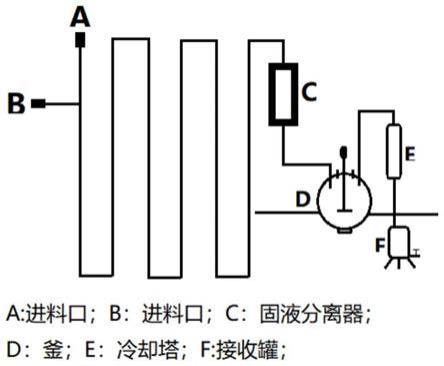
图3是根据本发明的微通道反应的工艺流程图。
具体实施方式
[0034]
需要说明的是,在本技术中,化合物1、化合物2、化合物3的结构式和名称如下:
[0035][0036]
发明人发现,基于化合物2生产化合物3的工艺路线是目前的主流生产路线。由于化合物2在乙醚和四氢呋喃中溶解性极差,而化合物3在反应溶剂中溶解性较好,如果要降低固液配比,使反应体积更适合工业化,那就会出现非均相的反应情况,生成的产品大部分溶解在溶液中,进一步与已溶解的碘反应生成副产物二碘化物,这也是为何目前的反应无法避免二碘化物生成的原因之一。要解决目前的技术问题,需要根据反应的原理和物质的理化性质进行设计。
[0037][0038]
传统釜式反应的弊端之一就是过度反应问题,例如,当化合物a和b反应生成c和d,如果c或d还有可以继续和a或b反应的位点,那么在反应条件缺少选择性的情况下,就无法避免地发生过度反应。在上面这个反应中,由于碘是过量投料的,反应过程中过量的碘会继续与产物即化合物3反应,生成二碘代物。文献tetrahedron letters,vol.24,no.l5,pp 1605-1608,1983,对该反应的机理进行了详细描述:
[0039][0040]
其中,化合物(1)被碘氧化为重氮化合物(2),化合物(2)在碘正离子的加成作用下生成中间态(3),中间态(3)放出氮气变为中间体(4),中间态(4)经质子转移消除变为化合物(6)或经质子加成变为化合物(5),中间态(4)发生质子转移生成化合物(6)和(5)的过程属于竞争反应,在碘过量的情况下无法避免这种竞争,且这些反应属于快速反应,难以通过参数对反应进行控制。
[0041]
实验显示,在釜式反应中,如果通过控制碘的用量,将碘的用量降低至1.0eq,则原料即化合物2就会出现剩余,即化合物2的转化率低,不能完全转化成化合物3。另外,反应在中间态(4)发生质子转移生成化合物(6)和(5)的过程属于竞争反应,由于无法控制碘和化合物2的分布,所以釜式反应就不可避免地产生副产物。
[0042]
要改变上述反应的选择性,控制过度反应是必要的,而微通道反应器则可以解决这个问题,通过特定比例的碘溶液和化合物2的匀速定量混合反应,则可以实现特定转化率的反应控制。
[0043]
具体而言,参见图3,通过如下流程实现化合物3的微通道反应生产:
[0044]
将化合物2溶于溶剂中,配制成浓度浆料a;将碘、四甲基胍溶于溶剂中,配制成浓度溶液b,连接微通道反应器,分别将物料a和物料b通过加料泵按照一定速率向微通道反应器进料,使得化合物2、碘、四甲基胍保持一定的固定物料摩尔配比进行反应,反应属于快反应,在60s内即可完成,微通道路径避免了试剂的过度和重复反应,可以较低的碘过量和仅产生少量二碘化物副产物的情况下,实施上述化合物2到化合物3的反应。
[0045]
反应结束后,将反应液固液分离,液体进入反应釜,加入淬灭剂(亚硫酸钠、硫代硫酸钠、焦亚硫酸钠)淬灭反应,有机相蒸馏浓缩,回收溶剂;浓缩剩余物加水洗涤,除去残留的少量无机盐,得到化合物3的粗品,粗品经乙醇水混合溶液重结晶,得到化合物3纯化品,纯化品纯度>98.0%。
[0046]
发明人经过反复试验和不断优化工艺,获得了稳定可控的工艺参数,具体包括:
[0047]
在根据本发明的方法的一个实施方案中,上述流程所述配制碘、四甲基胍溶液和配制化合物2浆料所用的溶剂包括甲基叔丁基甲醚、四氢呋喃中的一种或两种混合液。
[0048]
在根据本发明的方法的一个实施方案中,上述流程所述的化合物2、碘、四甲基胍的进料动态物料配比为1.0eq:2.0~3.0eq:2.0~5.0eq。
[0049]
在根据本发明的方法的一个实施方案中,上述流程所述的淬灭剂选择亚硫酸钠、硫代硫酸钠、焦亚硫酸钠的水溶液中的一种或多种。
[0050]
在根据本发明的方法的一个实施方案中,上述流程所述加热转化的混合溶剂指的是乙醇和水的混合溶剂,其中乙醇和水的比例为1:1~10:1(重量比),混合溶剂的用量为化合物2的5~12倍(重量比)。
[0051]
在根据本发明的方法的一个实施方案中,上述流程所述加热转化的温度为70℃~混合溶剂回流温度,加热转化时间为1.0h~2.0h。
[0052]
下文中,将参照实施例,更具体地描述本发明的各种实施方式,使得本领域的普通技术人员能够更好地理解本发明及其优点。需要说明的是,以下实施例对本发明技术方案作进一步非限制性的详细说明,其不应被认为是对本发明范围的限制,而只是本发明的示例性说明及典型代表。
[0053]
在这些实例中所用试剂,除特别说明的以外,均为化学纯试剂,供应商为福晨(天津)化学试剂有限公司,反应设备为高通量微通道反应器,小试及中试规模中采用通用型布氏漏斗进行过滤,生产批量采用离心机(lb600型)进行离心过滤;干燥设备采用真空烘箱(dzf-6020型)或双锥(fzg-8型)进行干燥。
[0054]
实施例1
[0055]
将3.0kg化合物2,加入25l四氢呋喃中,配成浓度为0.4kg/l浓度的浆料,记为浆料s;将6.35kg碘和5.8kg四甲基胍溶于25l四氢呋喃中,配成含碘1.0kg/l和四甲基胍浓度为2.0kg/l的四氢呋喃溶液,记为溶液r,将s料罐和r料罐分别连接微通道反应器的s进料口和r进料口,控制温度10~30℃,开启加料泵,调节进料速率,采用等速进料,物料在通道内反应结束后,反应液进入固液分离器,分出固体,液体进入釜中收集。釜内反应液中加入20%
的硫代硫酸钠水溶液10l,搅拌,淬灭反应。蒸馏回收四氢呋喃,蒸馏残余物加水10l,搅拌,离心,得到淡黄色固体。所得固体加入30l乙醇水混合液(乙醇:水=6:1,重量比),搅拌升温回流1h,冷却至15~25℃,搅拌析晶2h,离心过滤,滤饼于60℃真空干燥4h,得到产品3.6kg,收率91.1%。
[0056]
实施例2
[0057]
将4.8kg化合物2,加入40l四氢呋喃中,配成浓度约为0.4kg/l浓度的浆料,记为浆料s;将8.06kg碘和5.48kg四甲基胍溶于40l四氢呋喃中,配成含碘0.80kg/l和四甲基胍浓度为1.20kg/l的四氢呋喃溶液,记为溶液r,将s料罐和r料罐分别连接微通道反应器的s进料口和r进料口,控制温度10~30℃,开启加料泵,调节进料速率,采用等速进料,物料在通道内反应结束后,反应液进入固液分离器,分出固体,液体进入釜中收集。釜内反应液中加入20%的硫代硫酸钠水溶液15l,搅拌,淬灭反应。蒸馏回收四氢呋喃,蒸馏残余物加水15l,搅拌,离心,得到淡黄色固体。所得固体加入45l乙醇水混合液(乙醇:水=6:1,重量比),搅拌升温回流1h,冷却至15~25℃,搅拌析晶2h,离心过滤,滤饼于60℃真空干燥4h,得到产品5.6kg,收率88.9%。
[0058]
实施例3
[0059]
将3.0kg化合物2,加入50l甲基叔丁基醚中,配成浓度为0.2kg/l浓度的浆料,记为浆料s;将6.35kg碘和5.8kg四甲基胍溶于25l四氢呋喃中,配成含碘1.0kg/l和四甲基胍浓度为2.0kg/l的四氢呋喃溶液,记为溶液r,将s料罐和r料罐分别连接微通道反应器的s进料口和r进料口,控制温度10~30℃,开启加料泵,调节进料速率,使s料的进料速率为r料的2倍,物料在通道内反应结束后,反应液进入固液分离器,分出固体,液体进入釜中收集。釜内反应液中加入20%的硫代硫酸钠水溶液10l,搅拌,淬灭反应。蒸馏回收四氢呋喃,蒸馏残余物加水10l,搅拌,离心,得到淡黄色固体。所得固体加入32l乙醇水混合液(乙醇:水=6:1,重量比),搅拌升温回流2h,冷却至15~20℃,搅拌析晶4h,离心过滤,滤饼于60℃真空干燥4h,得到产品3.4kg,收率86.0%。
[0060]
图1所示证实根据本发明的方法制备的醋酸阿比特龙关键中间体化合物3的核磁共振氢谱(h nmr)图与目标化合物的结构相符。
[0061]
图2示出了根据本发明的方法制备的醋酸阿比特龙关键中间体化合物3的化学纯度谱图。测试结果显示,化合物3纯度可达到99.0%以上。
[0062]
以上结合了优选实施方式对本技术进行了说明,不过这些实施方式仅是范例性的,仅起到说明性的作用。在此基础上,可以对本技术进行多种替换和改进,这些均落入本技术的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1