一种化合物及在制备NAMPT蛋白自噬降解剂中的应用
一种化合物及在制备nampt蛋白自噬降解剂中的应用
技术领域
1.本发明属于医药技术领域,具体地说,涉及一种化合物及在制备nampt蛋白自噬降解剂中的应用。
背景技术:
2.烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,nad)是细胞能量代谢途径中不可或缺的电子载体,包括糖酵解、三羧酸循环和氧化磷酸化等。相比于正常细胞,肿瘤细胞能量代谢活跃,对nad依赖性更强,因此,干扰和阻断nad的合成途径能够抑制肿瘤生长。哺乳动物主要依赖于以烟酰胺为底物的补救合成途径合成nad,烟酰胺磷酸核糖转移酶(nicotinamide phosphoribosyltransferase,nampt)是该条途径的限速酶。相比于正常细胞,肿瘤细胞中nampt表达量更高,临床数据也证实了肿瘤组织中的nampt处于高表达状态。与正常细胞相比,肿瘤细胞更易被nampt抑制剂影响,其可阻断肿瘤细胞nad合成主要途径,继而诱导肿瘤细胞死亡。研究表明,nampt能够通过影响细胞能量代谢、dna转录和修复功能、细胞氧化还原稳态影响肿瘤细胞的增殖和分化;nampt可分泌至细胞外,通过影响巨噬细胞m1/m2型比例、髓源性抑制细胞(myeloid-derived suppressor cells,mdsc)动员、中性粒细胞表达发挥类细胞因子作用,影响免疫微环境。因此nampt是抗肿瘤研究中的一个重要靶点。
3.近年来,靶向蛋白降解技术发展,研究较为成熟的技术主要为蛋白水解靶向嵌合分子(proteolysis-targeting chimeric,protac),其可同时结合目的蛋白和e3连接酶,然后利用泛素-蛋白酶体途径降解靶蛋白。但protac技术也存在一些局限性,比如:pprotac依赖特定的e3连接酶和泛素-蛋白酶体途径,会限制其在某些细胞或具有蛋白酶体抗性的蛋白中的应用,也有可能导致耐药性的产生;而且protac主要降解可溶性蛋白,难以降解胞外蛋白和多聚蛋白。真核生物存在两条主要的蛋白降解途径,即泛素-蛋白酶体途径和溶酶体介导的自噬途径。为了能够克服现有的protac技术的不足,进一步拓展胞内蛋白靶向降解的方式,可开发基于自噬溶酶体途径的蛋白降解新技术。
4.受protac药物设计原理的启发,设想是否可以通过嵌合体药物设计策略实现对目标蛋白的自噬降解:将nampt蛋白和lc3蛋白通过嵌合分子拉近促进靶蛋白向溶酶体靠近,通过溶酶体介导的自噬诱导靶蛋白降解,从而开发基于自噬溶酶体途径的靶蛋白降解技术。
5.关于本发明的nampt蛋白自噬降解剂,目前还未见文献报道。
技术实现要素:
6.本发明的第一个目的是提供一种可作为nampt蛋白自噬降解剂的化合物。
7.本发明的第二个目的是提供一种所述化合物在制备nampt蛋白自噬降解剂中的应用。
8.本发明的第三个目的是提供一种所述化合物在制备nampt蛋白抑制剂中的应用。
9.本发明的第四个目的是提供一种所述化合物在制备治疗nampt介导的肿瘤的药物中的应用。
10.为了实现上述目的,本发明采用的技术方案如下:
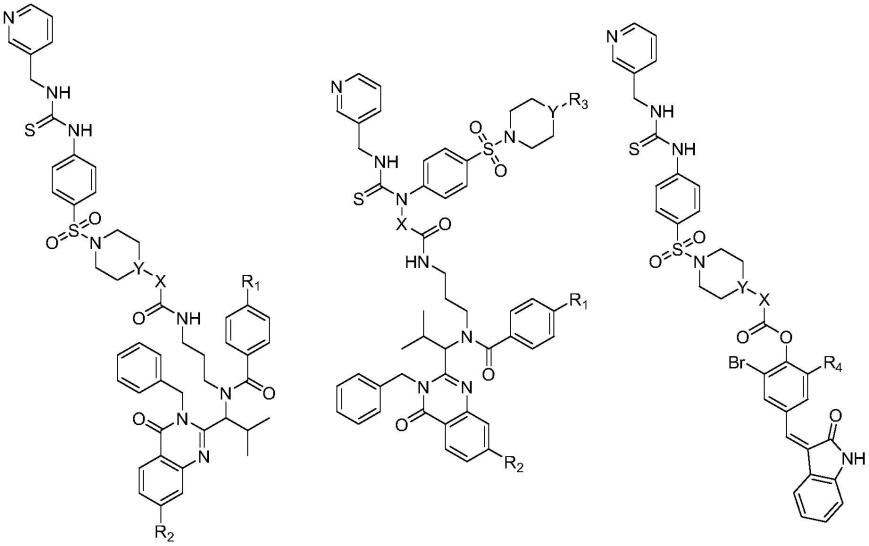
11.本发明的第一方面提供了一种化合物或其药用盐,结构如下所示:
[0012][0013]
其中,y选自n、ch;
[0014]
x选自-(ch2)n-、-(ch2ch2o)nch2ch
2-;n选自1~10的整数;
[0015]
r1选自c1~c5支链烷基、c1~c5直链烷基;
[0016]
r2选自氟、氯、溴;
[0017]
r3选自氢、c1~c5支链烷基、c1~c5直链烷基;
[0018]
r4选自氟、氯、溴、羟基。
[0019]
较优选的,所述化合物选自以下化合物的一种:
[0020][0021]
其中,x选自-(ch2)n-、-(ch2ch2o)nch2ch
2-;n选自1~10的整数(即1、2、3、4、5、6、7、8、9、10);
[0022]
r1选自c1~c5支链烷基、c1~c5直链烷基;
[0023]
r2选自氟、氯、溴;
[0024]
r4选自氟、氯、溴、羟基。
[0025]
所述化合物中,r1选自甲基、乙基、异丙基。
[0026]
最优选的,所述化合物选自以下结构的一种:
[0027]
[0028][0029]
所述药用盐包括通式的化合物与下列酸形成的酸加成盐:盐酸、氢溴酸、硫酸、乳酸、柠檬酸、磷酸、甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸、酒石酸、丙酮酸、乙酸、马来酸或琥珀酸、富马酸、水杨酸、苯基乙酸、杏仁酸。
[0030]
本发明的第二方面提供了一种所述化合物或其药用盐在制备nampt蛋白自噬降解剂中的应用。
[0031]
本发明的第三个方面提供了一种所述化合物或其药用盐在制备nampt蛋白抑制剂中的应用。
[0032]
nampt蛋白自噬降解剂可同时与nampt蛋白和自噬关键蛋白lc3结合,将nampt蛋白传送至溶酶体,诱导nampt蛋白降解,从而降低细胞内nampt含量,而nampt蛋白抑制剂主要通过小分子化合物与nampt蛋白相结合,占据其活性口袋从而抑制蛋白的相关活性来起作用。
[0033]
本发明的第四个方面提供了一种所述化合物或其药用盐在制备治疗nampt介导的肿瘤的药物中的应用。
[0034]
所述nampt介导的肿瘤选自肺癌、肝癌、肾癌、非小细胞肺癌、前列腺癌、甲状腺癌、皮肤癌、胰腺癌、卵巢癌、乳腺癌、膀胱癌、骨髓增生异常综合症、淋巴瘤、食管癌、胃肠道癌、骨肉瘤、中枢或外周神经系统的肿瘤。
[0035]
本发明的第五个方面提供了一种所述化合物或其药用盐在制备激活免疫系统的药物中的应用。
[0036]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0037]
本发明实验结果表明:本发明制备的化合物表现出良好的nampt酶抑制活性,能通过自噬降解途径下调细胞内nampt蛋白水平,实现对肿瘤的抑制,具有一定的抗肿瘤活性,可以应用于nampt介导的肿瘤疾病。
[0038]
本发明的化合物作为首次报道的nampt自噬降解剂,具有进一步的开发和研究价值。
[0039]
本发明化合物的制备方法简便,可重复性好,得率高。
附图说明
[0040]
图1是本发明化合物作用于a2780细胞48h后细胞内nampt蛋白含量的影响示意图。
[0041]
图2是a2780细胞经自噬抑制剂或nampt抑制剂预处理12h后,与化合物3或化合物11共孵育48h后细胞内nampt蛋白水平示意图。
[0042]
图3是化合物3作用于atg7敲低的a2780细胞48h后,细胞内nampt的含量变化示意图。
[0043]
图4是本发明化合物作用于a2780细胞24h后,细胞内nad的含量变化示意图。
具体实施方式
[0044]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0045]
在本发明中,下列实施例中所使用的试剂和原料均可通过商业途径购买获得。化学原料从毕得、泰坦、希恩斯等公司购买;细胞及生物材料从源叶生物、陶素生物等公司购买。
[0046]
以下实施例所涉化合物的1h nmr,
13
c nmr和ms数据详见表1。表1中序号1-12为化
合物编号,不仅一一对应于表1中的序号,同时也对应下面的实施例1-12制备的具体化合物。
[0047]
表1
[0048]
[0049]
[0050]
[0051]
[0052]
[0053][0054]
实施例1
[0055]
化合物1的制备方法如下:
[0056]
步骤a-e.(化合物n6)参照申请号为202010255728.4的专利申请合成。
[0057][0058]
步骤f.7-(4-((4-(3-(吡啶-3-基甲基)硫脲基)苯基)磺酰基)哌嗪-1-基)庚酸叔丁酯(n7-1)
[0059][0060]
将化合物n6(500mg,1.3mmol)和l1(338mg,1.3mmol)溶于干燥的dcm(20ml)中,缓慢滴加dipea(335mg,2.6mmol),随后在60℃条件下搅拌2h。tlc监测反应结束后,减压蒸除反应溶剂,剩余物经硅胶柱层析(dcm/meoh=100/1)分离纯化,得到白色固体n7-1(512mg,产率68%)。1h nmr(600mhz,dmso-d6)δ:10.17(s,1h),8.66(s,1h),8.59(s,1h),8.49(d,j=4.7hz,1h),7.82(d,j=8.6hz,2h),7.78(d,j=7.9hz,1h),7.67(d,j=8.6hz,2h),7.40(dd,j=7.8,4.9hz,1h),4.81(d,j=5.5hz,2h),2.87(s,4h),2.41(s,4h),2.24(s,2h),1.43-1.47(m,2h),1.38(s,9h),1.31-1.37(m,2h),1.17-1.27(m,6h).
[0061]
步骤g.(r)-n-(1-(3-苄基-7-氯-4-氧代-3,4-二氢喹唑啉-2-基)-2-甲基丙基)-4-甲基-n-(3-(7-(4-((4-(3-(吡啶-3-基甲基)硫脲基)苯基)磺酰基)哌嗪-1-基)庚酰胺基)丙基)苯甲酰胺(1)
[0062][0063]
将n7-1(500mg,0.87mmol)溶于含有20%三氟乙酸的二氯甲烷溶液(5ml)中,室温下搅拌2h,反应液浓缩得到n8-1(451mg,100%)。依次将中间体n8-1(519mg,1.0mmol)、化合物ispinesib(516mg,1.0mmol)和hatu(570mg,1.5mmol)溶于干燥的dmf(10ml)中,室温搅拌1h。tlc监测反应完全后,将反应液倒入冰水中(30ml),有大量白色固体析出,减压抽滤,滤饼在50℃条件下烘干至恒重,无需进一步纯化,获得白色固体产物化合物1(360mg,产率36%)。1h nmr(600mhz,dmso-d6)δ:10.14(s,1h),8.63(s,1h),8.57(d,j=1.7hz,1h),8.47(dd,j=4.8,1.3hz,1h),8.22(d,j=8.6hz,1h),7.81(d,j=8.6hz,2h),7.75-7.78(m,2h),7.63-7.67(m,3h),7.34-7.38(m,3h),7.28-7.33(m,2h),7.18-7.26(m,6h),5.87(d,j=16.4hz,1h),5.53(d,j=10.4hz,1h),4.80(d,j=5.5hz,2h),3.17-3.26(m,2h),2.66-2.75(m,1h),2.34-2.50(m,4h),2.31(s,3h),2.16-2.27(m,2h),1.68-1.79(m,2h),1.21-1.35(m,10h),1.05-1.17(m,5h),0.89(d,j=6.8hz,3h),0.79-0.86(m,2h),0.47(d,j=6.2hz,3h);
13
c nmr(150mhz,dmso-d6)δ:185.98,177.24,176.85,166.33,160.45,154.11,153.40,152.37,149.25,144.70,143.88,141.87,140.51,139.37,138.96,134.09,133.87,133.62,133.22,132.65,131.86,131.60,131.07,128.66,126.76,124.27,99.99,64.21,66.75,50.95,50.39,49.92,47.67,40.92,40.43,35.39,34.16,33.71,33.57,31.65,30.24,26.11,24.71,23.36;hrms(esi,positive)m/z calcd for c
54h65
cln9o5s2[m+h]
+
:1018.4233;found 1018.4260。
[0064]
实施例2
[0065]
化合物2的制备方法如下:
[0066][0067]
将实施例1中步骤f的原料l1替换为l2(摩尔用量与实施例1相同),其他与实施例1
相同,从而得到化合物2。
[0068]
实施例3
[0069]
化合物3的制备方法如下:
[0070][0071]
将实施例1中步骤f的原料l1替换为l3(摩尔用量与实施例1相同),其他与实施例1相同,从而得到化合物3。
[0072]
实施例4
[0073]
化合物4的制备方法如下:
[0074][0075]
将实施例1中步骤f的原料l1替换为l4(摩尔用量与实施例1相同),其他与实施例1相同,从而得到化合物4。
[0076]
实施例5
[0077]
化合物5的制备方法如下:
[0078]
[0079]
将实施例1中步骤f的原料l1替换为l5(摩尔用量与实施例1相同),其他与实施例1相同,从而得到化合物5。
[0080]
实施例6
[0081]
化合物6的制备方法如下:
[0082][0083]
实施例1中步骤f的原料l1替换为l6(摩尔用量与实施例1相同),其他与实施例1相同,从而得到化合物6。
[0084]
实施例7
[0085]
化合物7的制备方法如下:
[0086][0087]
将实施例1中步骤f的原料l1替换为l7(摩尔用量与实施例1相同),其他与实施例1相同,从而得到化合物7。
[0088]
实施例8
[0089]
化合物8的制备方法如下:
[0090][0091]
将实施例1中步骤f的原料l1替换为l8(摩尔用量与实施例1相同),其他与实施例1
相同,从而得到化合物8。
[0092]
实施例9
[0093]
化合物9的制备方法如下:
[0094]
步骤a.5-(1-(4-(哌嗪-1-基磺酰基)苯基)-3-(吡啶-3-基甲基)硫脲基)戊酸叔丁酯(n7-9)
[0095][0096]
将化合物n6(500mg,1.3mmol)和l9(306mg,1.3mmol)溶于干燥的dcm(20ml)中,缓慢滴加dipea(335mg,2.6mmol),随后在60℃条件下搅拌12h。tlc监测反应结束后,将反应液减压蒸馏,硅胶柱层析(dcm/meoh=100/1)分离纯化,得到白色固体n7-9(399mg,产率56%,纯度93.9%)。1h nmr(600mhz,dmso-d6)δ:8.55(s,1h),8.46(d,j=3.6hz,1h),7.75(d,j=7.6hz,1h),7.59(t,j=5.6hz,1h),7.52(d,j=8.4hz,1h),7.37(dd,j=7.6,4.7hz,1h),6.9(s,j=8.3hz,2h),4.48(d,j=5.7hz,2h),2.68-2.86(m,10h),2.15(t,j=6.7hz,2h),1.46-1.50(m,4h),1.38(s,9h),1.25-1.31(m,2h).
[0097]
步骤b.(r)-n-(1-(3-苄基-7-氯-4-氧代-3,4-二氢喹唑啉-2-基)-2-甲基丙基)-4-甲基-n-(3-(5-(1-(4-(哌嗪-1-基磺酰基)苯基)-3-(吡啶-3-基甲基)硫脲基)戊酰胺基)丙基)苯甲酰胺(9)
[0098][0099]
将n7-9(350mg,0.64mmol)溶于含有20%三氟乙酸的二氯甲烷溶液(5ml)中,室温下搅拌2h,反应液浓缩得到n8-9(314mg,100%)。将化合物n8-9(519mg,1.0mmol)、化合物ispinesib(516mg,1.0mmol)和hatu(570mg,1.5mmol)溶于干燥的dmf(10ml)中,室温搅拌1h。tlc监测反应完全后,将反应液倒入冰水中(30ml),乙酸乙酯萃取(15ml
×
3),将有机层合并后,用饱和nacl溶液(30ml)洗涤3次,无水硫酸钠干燥,硅胶柱层析(dcm/meoh=100/1)分离纯化,获得白色固体产物9(288mg,产率29%,纯度96.0%)。1h nmr(600mhz,dmso-d6)δ:8.52(s,1h),8.43(d,j=4.6hz,1h),8.20(d,j=8.7hz,1h),7.68-7.80(m,2h),7.56-7.68(m,2h),7.50(d,j=8.3hz,2h),7.11-7.43(m,11h),6.86(d,j=8.5hz,2h),5.85(d,j=
16.1hz,1h),5.50(d,j=10.6hz,1h),5.31(t,j=4.8hz,1h),5.02(d,j=16.2hz,1h),4.46(d,j=5.3hz,2h),3.14-3.23(m,2h),2.76(s,10h),2.29(s,3h),1.87-2.05(m,2h),1.62-1.83(m,2h),1.26-1.40(m,6h),0.82-0.89(m,4h),0.44(d,j=6.2hz,3h);hrms(esi,positive)m/z calcd for c
52h61
cln9o5s2[m+h]
+
:990.3920;found 990.3897.
[0100]
实施例10
[0101]
化合物10的制备方法如下:
[0102][0103][0104]
将实施例9中步骤f的原料l9替换为l10(摩尔用量与实施例9相同),其他与实施例9相同,从而得到化合物10。
[0105]
实施例11
[0106]
化合物11的制备方法如下:
[0107][0108]
将实施例9中步骤f的原料l9替换为l7(摩尔用量与实施例9相同),其他与实施例9相同,从而得到化合物11。
[0109]
实施例12
[0110]
化合物12的合成
[0111][0112]
依次将中间体n8-1(519mg,1.0mmol)、化合物(z)-3-(3,5-二溴-4-羟基苄叉)吲哚-2-酮(392mg,1.0mmol)、cdi(243mg,1.5mmol)和dmap(183mg,1.5mmol)溶于干燥的二氯甲烷(10ml)中,室温搅拌1h。tlc监测反应完全后,反应液浓缩柱层析,获得白色固体产物12(224mg,产率25%)。
[0113]
实施例13
[0114]
本发明化合物对nampt抑制活性测试
[0115]
一、实验方法
[0116]
实验材料:nampt蛋白,tris-hcl(ph 7.5),mgcl2,烟酰胺(nam),磷酸核糖焦磷酸(prpp),atp,乙醇脱氢酶,乙醇,二硫苏糖醇(dtt),牛血清白蛋白(bsa),吐温20(tween 20)。
[0117]
仪器:gen5操作软件,bioteck synergy2多功能酶标仪
[0118]
样品配置:样品测试液配置:50mm tris hcl(ph 7.5),12.5mm mgcl2,0.4mm prpp,2mm atp,30μg/ml乙醇脱氢酶,1.5%乙醇,2mm dtt,0.02%bsa,2μg/ml nampt。
[0119]
实验方法:
[0120]
步骤a.在96孔板中加入不同浓度的化合物的dmso溶液0.5μl和样品测试液(20μ
l);
[0121]
步骤b.室温孵育5min后,加入0.2μm的nam溶液(4.5μl);
[0122]
步骤c.37℃下孵育15min后,在95℃下加热1min终止反应;
[0123]
步骤d.反应液放置冰上冷却,加入20%苯乙酮(10μl)、2n的koh溶液(10μl),涡旋在冰上放置2min,加入88%甲酸(45μl),在37℃下孵育10min;
[0124]
步骤e.使用酶标仪测试激发波长为382nm、发射波长为445nm处的荧光值。
[0125]
步骤f.根据公式计算抑制率,利用graphpad软件拟合抑制率与化合物浓度相关的曲线,模拟得出ic
50
值。
[0126]
实验结果:本发明化合物的ic
50
值如表2所示,测试化合物表现出中等到优秀的抑制活性。
[0127]
表2、目标化合物对nampt的抑制活性(单位nmol/l)
[0128][0129]
实施例14
[0130]
本发明化合物的体外抗肿瘤活性测试(ic
50
)
[0131]
实验方法
[0132]
实验材料:细胞株(a2780)、培养基(rmpi高糖培养基)、cck8测试盒、胎牛血清(fbs)、双抗、磷酸盐缓冲液(pbs)、谷氨酰胺。
[0133]
仪器:gen5操作软件,bioteck synergy2多功能酶标仪。
[0134]
样品配置:培养基配置:90%的1640基础培养基+10%fbs和1%双抗+1%谷氨酰胺。
[0135]
实验方法:
[0136]
步骤a.接种6
×
103个/孔细胞(100μl)于96孔板中,周围加入pbs(100μl);
[0137]
步骤b.放置于细胞孵育箱中37℃、5%co2条件下培养24h;
[0138]
步骤c.加入使用培养基配置的不同浓度的待测化合物(100μl),并设置三复孔;
[0139]
步骤d.放置于细胞孵育箱中培养72h,弃培养基,加入含有10%cck8的培养基(100μl);
[0140]
步骤e.在37℃下孵育40min后,用酶标仪测试样品在452nm处的od值;
[0141]
步骤f.利用graphpad模拟计算其ic
50
。
[0142]
实验结果:本发明化合物对a2780肿瘤细胞半数抑制浓度ic
50
值如表3所示,测试结
果显示,大部分化合物表现出中等到优秀的抗肿瘤活性。
[0143]
表3、目标化合物对肿瘤细胞的半数抑制浓度ic
50
(单位μmol/l)
[0144][0145]
实施例15
[0146]
本发明化合物对细胞内外nampt蛋白含量的影响
[0147]
一、实验方法
[0148]
(1)实验材料:蛋白强裂解液,蛋白水解酶抑制剂,磷酸酶抑制剂,pbs,bca试剂盒,蛋白上样缓冲液,双色预染蛋白marker,tris/甘氨酸/sds电泳缓冲液,转膜缓冲液,pvdf膜,牛血清白蛋白(bsa),tbst,rabbit anti-visfatin antibody(abcam,ab236873),rabbit anti-gapdh antibody(abcam,ab181602)和goat anti-rabbit igg h&l(alexa680)(abcam,ab175773)。
[0149]
(2)仪器:gen5操作软件,bioteck synergy2多功能酶标仪,电泳仪电源,小型垂直电泳仪,快速转膜系统,licor odyssey双色红外激光成像仪。
[0150]
(3)样品配置:蛋白裂解液混合物的成分为蛋白强裂解液和蛋白水解酶抑制剂、磷酸酶抑制剂,体积比为100:1:1。
[0151]
(4)实验方法:
[0152]
步骤a.将a2780细胞以5
×
105个/孔的密度接种于6孔板中(2ml),培养24h,之后加入相应浓度的药物继续培养72h;
[0153]
步骤b.收集细胞,用4℃预冷的pbs润洗一遍,加入含有蛋白水解酶抑制剂和磷酸酶抑制剂的细胞强裂解液(60μl),冰浴条件下孵育30min;
[0154]
步骤c.收集细胞裂解混合物,放置于离心管中,涡旋,在冰浴中静置5min,重复三次;
[0155]
步骤d.将混合液置于低温高速离心机中离心,在4℃、1.2
×
104rpm条件下离心15min;
[0156]
步骤f.取上清液(40μl)于离心管中,加入蛋白上样缓冲液(10μl),于100℃条件下孵育15min使蛋白变性;
[0157]
步骤g.取总蛋白(40μg)进行电泳(100v,2h);
[0158]
步骤h.在270ma条件下转膜约100min;
[0159]
步骤i.然后在5%的bsa溶液中室温封闭2h;
[0160]
步骤j.4℃孵一抗(1:1000)过夜;
[0161]
步骤k.回收后用tbst洗三次,每次5min;
[0162]
步骤l.室温孵二抗1h;
[0163]
步骤m.回收后用tbst洗三次,每次5min;
[0164]
步骤n.在odyssey双色红外激光成像系统扫描显影,gapdh作为内参。
[0165]
二、实验结果
[0166]
本发明化合物能够降解细胞内nampt蛋白,如图1所示,图1是本发明化合物作用于a2780细胞48h后细胞内nampt蛋白含量的影响示意图,说明大部分化合物能够有效降解a2780细胞中的nampt蛋白。
[0167]
实施例16
[0168]
本发明化合物的作用机制
[0169]
一、实验方法
[0170]
(1)实验材料:慢病毒颗粒(购于汉恒生物),polybrene,puromycin。
[0171]
(2)实验方法:
[0172]
步骤a.将a2780细胞以5
×
105个/孔的密度接种于6孔板中(2ml),培养24h;
[0173]
步骤b.弃培养基,加入含有慢病毒颗粒和polybrene的培养基(1ml),培养4h;
[0174]
步骤c.加入纯1640培养基(1ml),继续培养48h;
[0175]
步骤d.更换培养基为含有puromycin的培养基,孵育24h;
[0176]
步骤e.裂解细胞提取蛋白,进行western blot蛋白含量测试。
[0177]
二、实验结果
[0178]
如图2所示,共孵育后,nampt蛋白水平也有不同程度的恢复,加入nampt抑制剂fk866或ms2与化合物3或11竞争nampt蛋白的活性口袋后,细胞自噬被阻断,nampt蛋白水平恢复,表明化合物3和11通过溶酶体介导的自噬引起nampt蛋白水平下降。
[0179]
利用慢病毒颗粒感染a2780细胞,敲低自噬相关基因atg7从而阻断自噬过程,如图3所示,从图中可以看出,化合物3不能够降解nampt蛋白,表明化合物3通过溶酶体介导的自噬引起nampt蛋白水平下降。
[0180]
图2是a2780细胞经自噬抑制剂或nampt抑制剂预处理12h后,与化合物3(a)或化合物11(b)共孵育48h后细胞内nampt蛋白水平示意图,说明自噬抑制剂或nampt抑制剂能够挽救化合物3或11造成的降解,说明化合物是通过自噬途径引起的nampt蛋白降解。图3是化合物3作用于atg7敲低的a2780细胞48h后,细胞内nampt的含量变化示意图。
[0181]
实施例17
[0182]
本发明化合物对细胞内nad的影响
[0183]
一、实验方法
[0184]
(1)实验材料:1m的hclo4溶液,1m的k2co3溶液,tris-hcl(ph 7.5),乙醇,吩嗪硫酸乙酯(pes,phenazine ethosulfate),噻唑蓝,乙醇脱氢酶(adh,alcohol dehydrogenase)。
[0185]
(2)仪器:gen5操作软件,biotek synergy2多功能酶标仪。
[0186]
(3)样品配置:测试反应液成分为50mm的tris-hcl(ph7.5)、3%的乙醇、1.66mm吩
嗪硫酸乙酯、0.42mm噻唑蓝、90μg/ml乙酸脱氢酶。
[0187]
(4)实验方法:
[0188]
步骤a.将细胞铺板于96孔板中培养,当细胞融合至70-80%左右时,更换为无血清的培养基对细胞进行饥饿处理12h;
[0189]
步骤b.加入不同浓度的待测化合物并设置空白组,作用24h;
[0190]
步骤c.弃培养基,加入1n的hclo4溶液(50μl),作用30min以裂解细胞;
[0191]
步骤d.置于96孔离心机中,在1.2
×
104rpm条件下离心5min;
[0192]
步骤e.取上清液(40μl)于离心管内,加入1n的k2co3溶液(20μl),进行中和,冰浴20min;
[0193]
步骤f.反应液在1.2
×
104rpm条件下离心5min,取上清液(10μl)与反应液(90μl)混合,37℃下孵育40min;
[0194]
步骤g.测定570nm处的吸光度值,空白为不含乙醇脱氢酶的体系。
[0195]
二、实验结果
[0196]
nad下调是nampt蛋白活性下调的重要指标。当化合物与a2780细胞共孵育24h后,能明显降低细胞内nad水平,证明化合物降低了nampt蛋白的活性。如图4所示,图4是本发明化合物作用于a2780细胞24h后,细胞内nad的含量变化示意图。
[0197]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1