对二甲苯二磷酸四乙酯的合成方法与流程
1.本发明涉及有机合成技术领域,具体涉及一种对二甲苯二磷酸四乙酯的制备方法。
背景技术:
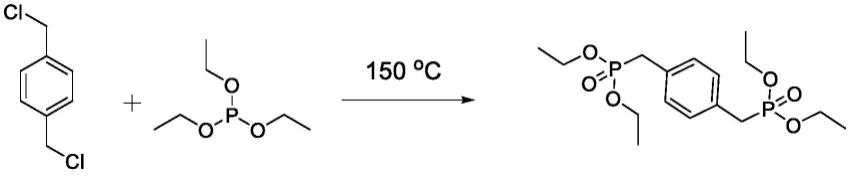
2.现有的对二甲苯二磷酸四乙酯的合成方法,以对二氯苄与亚磷酸三乙酯为原料,经150℃以上的高温回流制得,合成路线如下:
[0003][0004]
在对二甲苯二磷酸四乙酯的合成中,通常采用无溶剂反应,加热至亚磷酸三乙酯回流,温度需要达到150℃以上。高温对工业设备要求高,而且这对于工业生产来说很难实现,并且具有较大的危险性,难以实现工业化生产。
技术实现要素:
[0005]
有鉴于此,本发明提供一种无需高温,安全稳定的对二甲苯二磷酸四乙酯的合成方法。
[0006]
为解决上述技术问题,本发明采用以下技术方案:
[0007]
根据本发明实施例的对二甲苯二磷酸四乙酯的合成方法,包括如下步骤:
[0008]
以亚磷酸二乙酯作为底物,与亲核试剂进行脱氢反应;
[0009]
在反应液中继续加入对二卤苄发生取代反应,得到所述对二甲苯二磷酸四乙酯。
[0010]
进一步地,所述亲核试剂为选自氢化钠、乙醇钠、甲醇钠、乙醇钾、甲醇钾中的任意一种或多种。
[0011]
进一步地,所述对二卤苄为选自对二氯苄、对二溴苄和对二碘苄中的任意一种。
[0012]
进一步地,所述亚磷酸二乙酯:亲核试剂:对二卤苄的摩尔比为(2.0-3.0):(2.0-3.0):1。
[0013]
进一步地,所述脱氢反应以及所述取代反应在溶剂中进行,所述溶剂为选自乙醇、甲醇、二氯甲烷、乙酸乙酯、石油醚和四氢呋喃中的任意一种。
[0014]
更进一步地,所述脱氢反应具体包括:
[0015]
将所述亲核试剂加入所述溶剂中,并加热至所述亲核试剂完全溶解;
[0016]
向其中滴加所述亚磷酸二乙酯,以进行所述脱氢反应。
[0017]
进一步地,滴加所述亚磷酸二乙酯之后进行0.5-4小时的所述脱氢反应。
[0018]
进一步地,所述取代反应在升温回流条件下进行4-24小时。
[0019]
进一步地,还包括如下步骤:
[0020]
反应结束后,进行旋转蒸发去除所述溶剂,以得到初浓缩体系;
[0021]
采用分液法对所述初浓缩体系进行提取,得到有机相;
[0022]
对所述有机相进行浓缩,得到粗产物;
[0023]
对所述粗产物采用重结晶的纯化方法,得到成品。
[0024]
本发明的上述技术方案至少具有如下有益效果之一:
[0025]
根据本发明实施例的对二甲苯二磷酸四乙酯的合成方法,代替现有方法中的亚磷酸三乙酯而采用亚磷酸二乙酯作为底物,并首先使该底物发生脱氢反应之后与对二氯苄进行反应,避免了现有技术中的高温反应,降低了对生产设备的要求,同时增加了反应的安全性及可控性;
[0026]
进一步地,通过选用合适的溶剂,在溶剂中进行相应的回流反应,能够进一步大大降低反应温度,增加了反应的安全性及可控性。
具体实施方式
[0027]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例对本发明的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于所描述的本发明的实施例,本领域普通技术人员所获得的所有其他实施例,都属于本发明保护的范围。
[0028]
下面首先具体描述根据本发明实施例的对二甲苯二磷酸四乙酯的合成方法。
[0029]
根据本发明实施例的对二甲苯二磷酸四乙酯的合成方法,包括如下步骤:
[0030]
s1,以亚磷酸二乙酯作为底物,与亲核试剂进行脱氢反应。
[0031]
也就是说,首先,使亚磷酸二乙酯的磷位上的氢脱除,使其成为负离子团。
[0032]
具体而言,所述亲核试剂例如可以选自氢化钠、乙醇钠、甲醇钠、乙醇钾、甲醇钾中的任意一种或多种。
[0033]
更进一步地,为了降低反应温度,所述脱氢反应以及后续的取代反应在溶剂中进行。也就是说,在从最初开始使得底物在溶剂中进行脱氢反应,并继续在该反应体系中加入二卤苄完成后续的取代反应。
[0034]
优选地,所述溶剂可以为选自乙醇、甲醇、二氯甲烷、乙酸乙酯、石油醚和四氢呋喃中的任意一种。作为底物的亚磷酸二乙酯,以及亲核试剂、以及后续的对二卤苄都能很好地溶解在上述溶剂中,故而优选。
[0035]
作为一个示例,所述脱氢反应具体包括:
[0036]
将所述亲核试剂加入所述溶剂中,并加热至所述亲核试剂完全溶解;
[0037]
向其中滴加所述亚磷酸二乙酯,以进行所述脱氢反应。
[0038]
进一步地,滴加所述亚磷酸二乙酯之后进行0.5-4小时的所述脱氢反应。
[0039]
s2,在反应液中继续加入对二卤苄发生取代反应,得到所述对二甲苯二磷酸四乙酯。
[0040]
也就是说,在亚磷酸二乙酯脱氢之后,直接在反应体系中加入对二卤苄以发生取代反应,生成对二甲苯二磷酸四乙酯。
[0041]
其中,所述对二卤苄,例如可以选自对二氯苄、对二溴苄和对二碘苄中的任意一种。
[0042]
优选地,所述亚磷酸二乙酯:亲核试剂:对二卤苄的摩尔比为(2.0-3.0):(2.0-3.0):1。
[0043]
下面,结合具体实施例进一步详细说明根据本发明的对二甲苯二磷酸四乙酯的合成方法。
[0044]
实施例1对二甲苯二磷酸四乙酯的制备
[0045]
对二甲苯二磷酸四乙酯的化学结构式如下式所示:
[0046][0047]
向三口烧瓶中加入乙醇钠(16.33g,0.24mol)和乙醇,加热至乙醇钠溶清后滴加亚磷酸二乙酯(30.38g,0.22mol),滴加结束反应半小时后,分批加入对二氯苄(17.51g,0.1mol),加完后升温回流16小时。
[0048]
反应液经分液、萃取、浓缩等后处理得到粗品,经石油醚体系重结晶得到对二甲苯二磷酸四乙酯25.51g,收率67.43%,熔点73.6-75.5℃。
[0049]
对得到的产物进行核磁共振试验,数据如下:
[0050]
1h nmr(400mhz,cdcl3):δ=6.94(s,4h,4arh),3.0-3.04(m,4h,2ch2),4.05-4.08(m,8h,4ch2),1.10-1.14(m,12h,4ch3)。
[0051]
实施例2对二甲苯二磷酸四乙酯的制备
[0052]
向三口烧瓶中加入乙醇钠(16.33g,0.24mol)和乙醇,加热至乙醇钠溶清后滴加亚磷酸二乙酯(30.38g,0.22mol),滴加结束反应半小时后,分批加入对二溴苄(26.4g,0.1mol),加完后升温回流8小时。反应液经分液、萃取、浓缩等后处理得到粗品,经石油醚体系重结晶得到对二甲苯二磷酸四乙酯26.10g,收率69.0%,熔点73.5-75.9℃。
[0053]
实施例3对二甲苯二磷酸四乙酯的制备
[0054]
向三口烧瓶中加入乙醇钠(16.33g,0.24mol)和乙醇,加热至乙醇钠溶清后滴加亚磷酸二乙酯(30.38g,0.22mol),加入碘化钾(3g,0.02mol)催化,分批加入对二氯苄(17.51g,0.1mol)。加完后升温回流至点板原料基本消耗完。反应液经分液、萃取、浓缩等后处理得到粗品,经石油醚体系重结晶得到对二甲苯二磷酸四乙酯26.48g,收率69.98%,熔点73.8-75.2℃。
[0055]
实施例4对二甲苯二磷酸四乙酯的制备
[0056]
向三口烧瓶中加入乙醇钠(20.42g,0.3mol)和乙醇,加热至乙醇钠溶清后滴加亚磷酸二乙酯(30.38g,0.22mol),滴加结束反应半小时后,分批加入对二氯苄(17.51g,0.1mol)。加完后升温回流至点板原料基本消耗完。反应液经分液、萃取、浓缩等后处理得到粗品,经石油醚体系重结晶得到对二甲苯二磷酸四乙酯24.21g,收率63.99%,熔点73.6-75.4℃。
[0057]
实施例5对二甲苯二磷酸四乙酯的制备
[0058]
向三口瓶中加入乙醇钠(21.78g,0.32mol)和乙醇,加热至乙醇钠溶清后滴加亚磷酸二乙酯(41.43g,0.3mol),滴加结束反应半小时后,加入对二氯苄(17.51g,0.1mol)。加完后升温回流至点板原料基本消耗完。反应液经分液、萃取、浓缩等后处理得到粗品,经石油
醚体系重结晶得到对二甲苯二磷酸四乙酯29.67g,收率78.43%,熔点73.5-75.2℃
[0059]
实施例6对二甲苯二磷酸四乙酯的制备
[0060]
向三口瓶中加入乙醇钠(14.97g,0.22mol)和乙醇,加热至乙醇钠溶清后滴加亚磷酸二乙酯(34.52g,0.25mol),滴加结束反应半小时后,分批加入对二氯苄(17.51g,0.1mol)。加完后升温回流至点板原料基本消耗完。反应液经分液、萃取、浓缩等后处理得到粗品,经石油醚体系重结晶得到对二甲苯二磷酸四乙酯22.73g,收率60.1%,熔点74.6-75.8℃。
[0061]
实施例7对二甲苯二磷酸四乙酯的制备
[0062]
向三口瓶中加入乙醇钠(17.69g,0.26mol)和乙醇,加热至50℃溶清后滴加亚磷酸二乙酯(34.52g,0.25mol),分批加入对二氯苄(17.51g,0.1mol)。升温至回流反应8h,tlc监测反应原料消耗完,反应液经分液、萃取、浓缩等后处理得到粗品,经石油醚体系重结晶得到对二甲苯二磷酸四乙酯31.58g,收率83.46%,熔点73.4-75.8℃。
[0063]
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1