以甘蔗废糖蜜为原料合成荧光碳点的制备方法及其应用与流程
本发明涉及一种荧光碳纳米材料-碳点的制备及应用,即以甘蔗废糖蜜为原料,经高温裂解制备荧光碳点,并系统研究该碳点在细胞成像及日落黄的含量检测的应用,属于生物材料、生物医学和食品检测领域。
背景技术:
废糖蜜是一种黑色粘稠液体,是生产原糖和精糖的副产物。其主要成分为发酵性糖和非发酵性糖,含糖量约为45~50%。由于糖蜜具有较高的含糖量,因此常在农副产品加工、食品、建筑、能源化工和轻工等领域发挥着重要作用。在农副产品领域中,糖蜜可作为辅料,添加至饲料中,有助于牲畜补充能量和矿物质,促进消化吸收,提高牲畜肉质和牛乳产量。在食品领域中,糖蜜与氨基化合物反应,可制备出亲水性良好,着色能力强,安全高效的食用色素。在建筑领域中,水泥中添加糖蜜,可提高水泥的流动性、凝固时间、力学强度等性能。在能源化工领域中,糖蜜通过发酵作用产生乙醇、丁醇等醇类,部分替代汽油,实现能源可持续发展。此外,糖蜜在氨基酸(亮氨酸、谷氨酸、赖氨酸等)工业生产中具有广泛应用。在轻工领域中,掺杂少量糖蜜,利用糖蜜中的单糖和蔗糖与纤维素相结合,可提高纸质的伸张性和保湿性。由此可见,糖蜜作为添加剂被广泛应用于农副产品加工、食品、建筑、能源化工和轻工等领域。但是通过直接或间接地改性糖蜜,应用于生物医学领域中的研究和应用报道未见报道。
碳点是一类以碳为基本结构单元,尺寸小于20 nm,且具有良好亲水性和荧光性质的纳米材料。碳点的荧光性质依赖于激发波长,即发射光谱可随着激发波长的增加而发生明显红移。碳点具有良好的抗漂白性,在氙灯或激光共聚焦显微镜直接照射下,荧光强度基本无变化。与传统的金属量子点相比,碳点具有优异的生物相容性。基于碳点显著的特征,碳点可用于生物探针、药物载体及活体成像等研究。目前有很多学者对碳点进行了一系列的研究,如检索到以下专利:
申请号:201511004716.X;申请人:江南大学;发明名称:一种以米糠为碳源制备荧光碳点的方法;摘要:本发明公开了一种以米糠为碳源制备荧光碳点的方法。将米糠作为碳源,采用水热法制备出荧光碳点。该方法将米糠放入马弗炉中煅烧后,加入去离子水于反应釜内加热数小时。将反应液离心,经滤膜过滤,再用透析袋透析后即得碳点溶液。本发明所用原料源于自然资源,易于获取,且制备方法简单、绿色环保。所得碳点具有很好的水溶性,荧光强度高,可适用于生物标记、细胞成像等领域。
申请号:201511004717.4;申请人:江南大学;发明名称:一种基于柚子制备荧光碳点的方法;摘要:本发明公开了一种基于柚子制备荧光碳点的方法。将柚子作为碳源,采用水热法制备出荧光碳点。该方法将柚子汁置于反应釜内反应数小时后,将反应液离心,经滤膜过滤,透析后得碳点溶液。本发明所用原料源于自然资源,易于获取,且制备方法简单、绿色环保。所得碳点具有很好的水溶性,荧光强度高,可适用于生物标记、细胞成像等领域。
申请号:201410623730.7;申请人:山西大学;发明名称:一种绿色荧光碳点的制备方法及在细胞成像方面的应用;摘要:本发明提供了一种绿色荧光碳点的制备方法及在细胞成像方面的应用,属于荧光纳米材料的制备和应用领域。本发明具体的制备步骤如下:(1) 将花瓣粉末加入去离子水中,制得混合液;(2) 将 (1) 得到的混合液转移到水热反应釜中,进行水热反应;(3) 将 (2) 得到的产物经过离心、透析,最终得到绿色荧光碳点溶液。本发明将植物花瓣作为碳源,原料广泛易得,制备方法简单,成本低;而且制得的绿色荧光碳点水溶性好、稳定性强,可直接用于细胞成像等。
上述制备荧光碳点的专利的碳源依次为米糠、柚子及花瓣,但是未见以废糖蜜为碳源制备荧光碳点的文献报道。本发明采用的是广西地区优势---废糖蜜,充分利用废液资源,符合变废为宝的政策。废糖蜜是制糖行业的废液,价格低廉,在广西来源广泛。废糖蜜多应用于工业等其他领域,在生物医学和食品检测未见有专利报道和公开。本发明以废糖蜜制备荧光碳点,应用于生物医学领域和食品检测。废糖蜜与其他碳源相比,最大区别是糖蜜是废液,其次广西是制糖大省,来源广泛,制备方法虽然与其他的制备方法基本相似。
日落黄是一种食用着色剂,常用于饮料、糕点及膨化食品等,禁止用于肉类、水果及婴儿食品等。国家食品药品监督管理局对于日落黄在各种食品用量作出了明确规定(GB 2760-2011 《食品安全国家标准食品添加剂使用标准》)。在规定用量范围内,日落黄对人体无明显危害。但人体过多地摄入日落黄,可能造成儿童智商低下,影响儿童肝脏和肾脏等发育,干扰体内代谢,引起多种疾病。因此,严格监管日落黄的使用,有效地检测食品中日落黄的含量,具有重要的意义。
技术实现要素:
针对上述存在的问题,本发明提供一种以甘蔗废糖蜜为原料合成荧光碳点的制备方法及其应用,即以甘蔗废糖蜜为原料,经高温裂解制备稳定性高,生物相容性好,且价格低廉的荧光碳点。该荧光碳点具有良好的抗漂白性、光稳定性和荧光性质,可用于细胞成像。此外,该荧光碳点能有效地检测日落黄的含量,将为食品添加剂中日落黄含量的检测提供一种有效、快速的方法。
为了实现上述目的,本发明采用的技术方案如下:
以甘蔗废糖蜜为原料合成荧光碳点的制备方法,包括如下步骤:
(1)、称取4.0 -6.0g的甘蔗废糖蜜,置入聚四氟乙烯内衬的反应釜中,于240-260℃反应11-13 h后,冷却至室温,得到高温裂解后的甘蔗废糖蜜;
(2)、往高温裂解后的甘蔗废糖蜜中再加入4-6 mL去离子水,在80-120 Hz超声2-4 min,得到甘蔗废糖蜜溶液;所述的甘蔗废糖蜜是甘蔗蔗糖生产过程结晶分离白糖后剩余的黑色粘稠液体;
(3)、将甘蔗废糖蜜溶液通过0.22μm 水相滤膜,反复过滤三次,收集金黄色滤液;
(4)、将金黄色滤液溶液滴入200 mL的无水乙醇中进行沉淀,析出褐色固体,于6000 round/min的转速下离心分离5min,收集澄清溶液,减压除去无水乙醇后,再加入10 mL去离子水,得荧光碳点溶液,最后将荧光碳点溶液通过0.22μm水相滤膜,收集滤液,冻干,得荧光碳点,备用。
优选地,以甘蔗废糖蜜为原料合成荧光碳点的制备方法,包括如下步骤:
(1)、称取5.0g的甘蔗废糖蜜,置入聚四氟乙烯内衬的反应釜中,于250℃反应12 h后,冷却至室温,得到高温裂解后的甘蔗废糖蜜;
(2)、往高温裂解后的甘蔗废糖蜜中再加入5 mL去离子水,在100 Hz超声3 min,得到甘蔗废糖蜜溶液;所述的甘蔗废糖蜜是甘蔗蔗糖生产过程结晶分离白糖后剩余的黑色粘稠液体;
(3)、将甘蔗废糖蜜溶液通过0.22μm滤膜,反复过滤三次,收集金黄色滤液;
(4)、将金黄色滤液溶液滴入200 mL的无水乙醇中进行沉淀,析出褐色固体,于6000 round/min的转速下离心分离5min,收集澄清溶液,减压除去无水乙醇后,再加入10 mL去离子水,得荧光碳点溶液,最后将荧光碳点溶液通过0.22μm水相滤膜,收集滤液,冻干,得荧光碳点,备用。
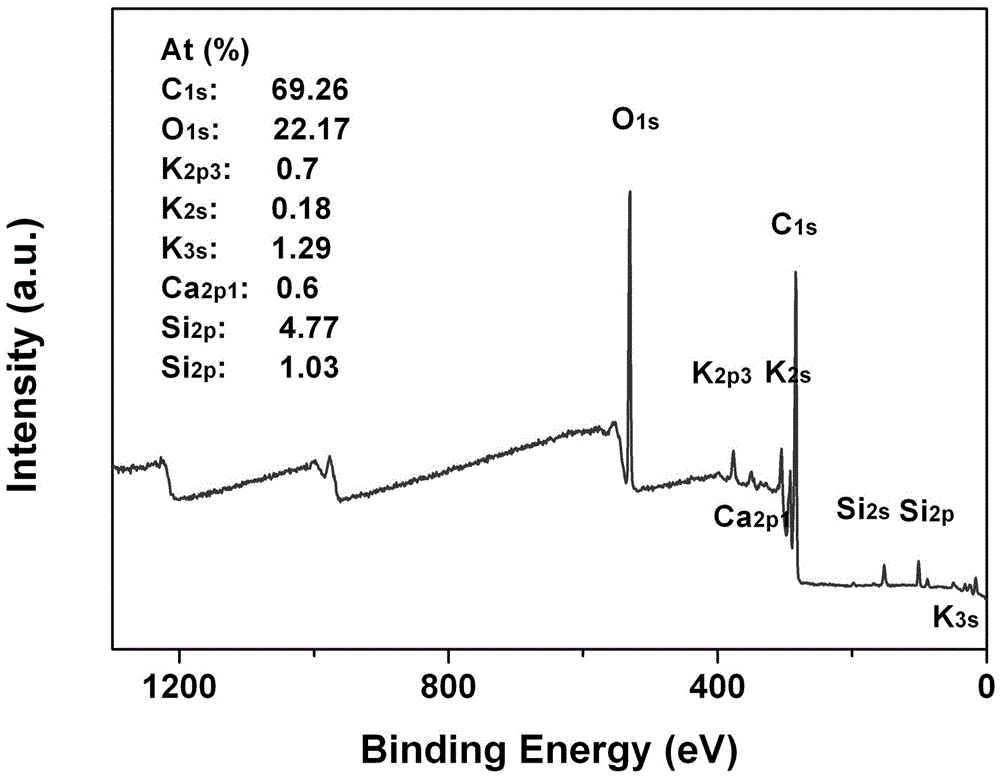
本发明所述的制备方法得到的荧光碳点,其X射线光电子能谱试验表明,碳点主要含有C、O元素,原子百分比分别69.26%,22.17%。
本发明所述的制备方法得到的荧光碳点,其FT-IR表明,3400 cm-1归属于-OH伸缩振动峰;2983cm-1归属于C-H伸缩振动峰;1565cm-1、1402cm-1归属于COO-非对称和对称伸缩振动峰;1120cm-1、1049cm-1归属于Si-O-Si对称伸缩振动峰。
本发明所述的制备方法得到的荧光碳点,其核磁共振氢谱1HNMR表明,化学位移在0.92~4.25 ppm归属于脂肪链上的氢;在6.17~8.47ppm归属于芳香化合物上的氢。
本发明所述的制备方法得到的荧光碳点,其核磁共振碳谱13CNMR表明,化学位移在8.48~68.45 ppm,179.76~182.35 ppm分别归属于脂肪族和芳香族上碳的特征吸收峰。
本发明所述的制备方法得到的荧光碳点,其透射电子显微镜TEM表明碳点尺寸为4.20 nm,晶格间距为0.23 nm。
本发明所述的制备方法得到的荧光碳点,其X射线衍射XRD表明碳点的2θ衍射峰主要位于24.64°,具有sp2碳的结构。
与现有技术相比,本发明的积极效果是
1、本发明的荧光碳点是利用甘蔗废糖蜜为原料,经过高温裂解制备。甘蔗废糖蜜原料价格廉价,合成工艺简易,可发挥广西地区优势(广西是制糖大省,来源广泛);废糖蜜与其他碳源相比,最大特点是废糖蜜是废液,来源广泛,本发明将废糖蜜制备为安全可靠的荧光碳点,应用于生物医学和食品检测,属于变废为宝的领域。比起采用柚子及花瓣作为荧光碳点,不仅来源广、成本低,制备过程工艺也较简单。
2、通过本发明的制备方法得到的荧光碳点稳定性高,生物相容性好,而且荧光特性优异;本发明的碳点可应用于在细胞内成像,并能有效检测食品添加剂中的日落黄含量。
3、本发明首次以废糖蜜为原料合成荧光碳点,拓展废糖蜜在生物医学及食品检测领域的应用。
4、本发明所述的制备方法得到的荧光碳点,其X射线光电子能谱试验表明,碳点主要含有C、O元素,原子百分比分别69.26%,22.17%;其FT-IR表明,3400 cm-1归属于-OH伸缩振动峰;2983cm-1归属于C-H伸缩振动峰;1565cm-1、1402cm-1归属于COO-非对称和对称伸缩振动峰;1120cm-1、1049cm-1归属于Si-O-Si对称伸缩振动峰;其核磁共振氢谱1HNMR表明,化学位移在0.92~4.25 ppm归属于脂肪链上的氢;在6.17~8.47ppm归属于芳香化合物上的氢;其核磁共振碳谱13CNMR表明,化学位移在8.48~68.45 ppm,179.76~182.35 ppm分别归属于脂肪族和芳香族上碳的特征吸收峰;其透射电子显微镜TEM表明碳点尺寸为4.20 nm,晶格间距为0.23 nm;其X射线衍射XRD表明碳点的2θ衍射峰主要位于24.64°,具有sp2碳的结构。
附图说明
图1为荧光碳点的X射线光电子能谱(XPS)图;
图2为荧光碳点的C1s图谱;
图3为荧光碳点的O1s图谱;
图4为荧光碳点的红外光谱(FT-IR)图;
图5为荧光碳点的1HNMR图;
图6为荧光碳点的13CNMR图;
图7为荧光碳点的透射电子显微镜(TEM)图;
图8为荧光碳点的X射线衍射(XRD)图;
图9为荧光碳点的热重分析仪(TGA)图谱;
图10为荧光碳点的紫外吸收光谱和荧光发射光谱;
图11为激发波长对碳点荧光光谱的影响;
图12为荧光碳点不同激发波长激发下的归一化荧光图;
图13为溶液pH对荧光碳点光谱性质的影响;
图14为NaCl溶液对荧光碳点光谱性质的影响;
图15为氨基酸对荧光碳点光谱性质的影响;
图16为金属离子对荧光碳点光谱性质的影响;
图17为荧光碳点在Fe3+不同浓度下荧光光谱;
图18为荧光碳点对Fe3+的线性关系;
图19为荧光碳点抗漂白性评价;
图20为荧光碳点自由基清除能力评价;
图21为荧光碳点的生物相容性评价;
图22为荧光碳点在细胞中成像,其溶血性质评价;
图23为碳点在MCF-7细胞内的明场照片;
图24为当激发波长为405 nm,荧光碳点在细胞内呈蓝色荧光;
图25为当激发波长为458nm,荧光碳点在细胞内呈绿色荧光;
图26为当激发波长为514 nm,荧光碳点在细胞内呈红色荧光;
图27为荧光碳点在日落黄不同浓度下的荧光光谱;
图28为荧光碳点对对日落黄的线性关系。
具体实施方式
实施例1
以甘蔗废糖蜜为原料合成荧光碳点的制备方法,包括如下步骤:
(1)、称取4.0 g的甘蔗废糖蜜,置入聚四氟乙烯内衬的反应釜中,于240℃反应13 h后,冷却至室温,得到高温裂解后的甘蔗废糖蜜;
(2)、往高温裂解后的甘蔗废糖蜜中再加入4mL去离子水,在80 Hz超声4 min,得到甘蔗废糖蜜溶液;所述的甘蔗废糖蜜是甘蔗蔗糖生产过程结晶分离白糖后剩余的黑色粘稠液体;
(3)、将甘蔗废糖蜜溶液通过0.22μm 水相滤膜,反复过滤三次,收集金黄色滤液;
(4)、将金黄色滤液溶液滴入200 mL的无水乙醇中进行沉淀,析出褐色固体,于6000 round/min的转速下离心分离5min,收集澄清溶液,减压除去无水乙醇后,再加入10 mL去离子水,得荧光碳点溶液,最后将荧光碳点溶液通过0.22μm水相滤膜,收集滤液,冻干,得荧光碳点,备用。
实施例2
以甘蔗废糖蜜为原料合成荧光碳点的制备方法,包括如下步骤:
(1)、称取5.0g的甘蔗废糖蜜,置入聚四氟乙烯内衬的反应釜中,于250℃反应12 h后,冷却至室温,得到高温裂解后的甘蔗废糖蜜;
(2)、往高温裂解后的甘蔗废糖蜜中再加入5 mL去离子水,在100 Hz超声3 min,得到甘蔗废糖蜜溶液;所述的甘蔗废糖蜜是甘蔗蔗糖生产过程结晶分离白糖后剩余的黑色粘稠液体;
(3)、将甘蔗废糖蜜溶液通过0.22μm水相滤膜,反复过滤三次,收集金黄色滤液;
(4)、将金黄色滤液溶液滴入200 mL的无水乙醇中进行沉淀,析出褐色固体,于6000 round/min的转速下离心分离5min,收集澄清溶液,减压除去无水乙醇后,再加入10 mL去离子水,得荧光碳点溶液,最后将荧光碳点溶液通过0.22μm水相滤膜,收集滤液,冻干,得荧光碳点,备用。
实施例3
以甘蔗废糖蜜为原料合成荧光碳点的制备方法,包括如下步骤:
(1)、称取6.0g的甘蔗废糖蜜,置入聚四氟乙烯内衬的反应釜中,于260℃反应11h后,冷却至室温,得到高温裂解后的甘蔗废糖蜜;
(2)、往高温裂解后的甘蔗废糖蜜中再加入6mL去离子水,在120Hz超声2 min,得到甘蔗废糖蜜溶液;所述的甘蔗废糖蜜是甘蔗蔗糖生产过程结晶分离白糖后以后剩余的废糖蜜;
(3)、将甘蔗废糖蜜溶液通过0.22μm水相滤膜,反复过滤三次,收集金黄色滤液;
(4)、将金黄色滤液溶液滴入200 mL的无水乙醇中进行沉淀,析出褐色固体,于6000 round/min的转速下离心分离5min,收集澄清溶液,减压除去无水乙醇后,再加入10 mL去离子水,得荧光碳点溶液,最后将荧光碳点溶液通过0.22μm水相滤膜,收集滤液,冻干,得荧光碳点,备用。
实施例2为最佳实施例,因此将实施例2所述的制备方法得到的荧光碳点,进行各种结构表征,如下表述。
将制备的碳点进行X射线光电子能谱(XPS)、红外光谱(FT-IR)、核磁共振(NMR)表征。XPS结果表明,碳点主要含有C、O元素,原子百分比分别69.26%,22.17%(图1)。此外,碳点中含有K、Ca、Si三种元素。C1s图谱表明,294.4 eV、291.7 eV归属于O-C=O;287.2eV、283.5eV分别归属于C-C、C-Si、C-O、C=C(图2)。O1s图谱表明O1s在530.4 eV有特征吸收峰(图3)。FT-IR表明,3400 cm-1归属于-OH伸缩振动峰;2983cm-1归属于C-H伸缩振动峰;1565cm-1、1402cm-1归属于COO-非对称和对称伸缩振动峰;1120cm-1、1049cm-1归属于Si-O-Si对称伸缩振动峰(图4)。1HNMR(核磁共振氢谱)表明,化学位移在0.92~4.25 ppm归属于脂肪链上的氢;在6.17~8.47ppm归属于芳香化合物上的氢(图5)。13CNMR(核磁共振碳谱)表明,化学位移在8.48~68.45 ppm,179.76~182.35 ppm分别归属于脂肪族和芳香族上碳的特征吸收峰(图6)。XPS、FT-IR和NMR结果表明碳点上含有羧基、羟基官能团。
通过透射电子显微镜(TEM)、X射线衍射(XRD)表征碳点的外观形貌和结构分析。TEM表明碳点尺寸为4.20 nm,晶格间距为0.23 nm(图7)。XRD谱表明,碳点的2θ衍射峰主要位于24.64°,具有sp2碳的结构(图8)。
利用热重分析仪(TGA)、紫外光谱(UV-Vis)、荧光光谱,研究碳点的热稳定性和光学性质。TGA结果表明,200℃时,由于碳点中的水蒸气蒸发,碳点总质量损失了8.56%,;460℃时,碳点总质量损失了35.32%;460~584℃,碳点恒重;900℃,碳点质量损失了83.59%(图9)。UV-Vis表明碳点溶液在260 nm和320 nm有明显的特征吸收峰(图10)。260 nm处特征吸收峰为C=C骨架组成的共轭双键中的π-π*跃迁的吸收峰位置;320 nm处吸收峰为羧基中C=O发生n-π*跃迁的吸收峰位置。以狭逢宽度为5 nm,激发波长为305 nm,在390 nm处出现碳点的荧光发射峰。在紫外灯(365 nm)照射下,呈蓝色荧光。配制0.25 mg/mL碳点水溶液,固定激发狭逢宽度为5 nm,改变激发波长(280~415 nm),在295~700 nm波长范围内扫描发射光谱。随着波长增大,碳点发射峰的最大吸收峰强度降低(图11)。将碳点发射峰归一化,结果表明碳点发射峰发生了红移(图12)。
荧光碳点的荧光性质表征:
a.配制不同pH水溶液(pH范围为3~12),碳点最终浓度为0.25mg/mL,在荧光光度计上(激发波长λex=309 nm)测定315-580 nm波长范围内发射光谱(图13)。
b.配制不同浓度的氯化钠(NaCl)溶液,碳点最终浓度为0.25mg/mL,扫描碳点在315-580 nm波长范围的发射光谱(图14)。
c.配制10种常见氨基酸水溶液和谷胱甘肽溶液(最终浓度为500μM),碳点最终浓度为0.25mg/mL,在荧光光度计上(λex=309 nm)测定315-580 nm波长范围内发射光谱(图15)。
d.配制13种常见金属水溶液,碳点最终浓度为0.25mg/mL,金属离子浓度为500μM,在荧光光度计上(λex=309 nm)测定315-580 nm波长范围内发射光谱(图16)。
上述结果表明,碳点的荧光不受溶液pH、NaCl、氨基酸影响,稳定性好。金属离子对碳点荧光性质实验表明,Zn2+、K+、Ca2+、Mg2+、Cd2+、La3+、Pb2+、Mn2+、Co2+、Cr3+、Fe2+、Cu2+对碳点的荧光影响较小。其中,Fe3+能猝灭碳点的荧光。随着Fe3+浓度提高,碳点荧光强度降低(图17)。Fe3+浓度在5~100μM时,碳点对Fe3+具有线性关系,方程为:y=0.0035x-0.0263,根据检测限公式:LOD=3σ/S,计算出碳点对Fe3+的检测限为3.79μM(图18)。
光稳定性和自由基清除活性评价:
配制13个0.25 mg/mL碳点溶液,分别在UV灯(365 nm)连续3 h照射下,每隔15 min取一次样品,测定390 nm处荧光强度(λex=309 nm)(图19)。结果表明,碳点的荧光强度无明显变化,具有良好的光稳定性。
4 mL 甲醇溶解4.0 mg 1,1-二苯基-2-三硝基苯肼(DPPH),准确量取1.26 mL DPPH的甲醇溶液,加入58.74 mL甲醇。移取上述DPPH溶液3 mL,加入稀释后碳点溶液200μL,充分混合均匀后,DPPH最终浓度为50μM,碳点最终浓度为(0 ~2.0 mg/mL)静置90 min,测定UV 在517 nm处测定紫外吸收(图20)。实验结果表明,EC50(能使50% DPPH自由基猝灭的碳点有效浓度)为0.514 mg/mL,不易受自由基影响。
在体外评价荧光碳点的细胞毒性和成像:
乳腺癌细胞(MCF-7)以每孔10000的密度接种于96孔板,在37℃、CO2浓度为5%的恒温培养箱中培养24 h后,加入不同浓度的碳点溶液(0.4~4.0 mg/mL)共孵育24 h,以培养基RPMI-1640作为对照,然后加入水溶性四氮唑(WST-1),孵育4 h后,吸去培养基,加入DMSO,在酶标仪上摇晃2 min,波长为450nm测吸光值。WST结果表明,碳点浓度在0.4~4 mg/mL范围内,培养24 h后,MCF-7成活率均高于80%,表明碳点具有良好的生物相容性(图21)。
为了评价碳点对细胞膜破坏程度,本发明进行了溶血实验(图22)。用pH=7.4的磷酸缓冲溶液(PBS),加入至人血中,离心(1500 round/min,10 min),洗涤六次,用PBS稀释至人的红血球细胞(HRBC)浓度为4×108个/mL。加入200μ L含HRBC的PBS,600μL PBS和不同浓度的碳点溶液,以PBS和去离子水作为阴性和阳性对照,37℃下孵育2 h,离心(1500 round/min,10 min),取上层澄清溶液,利用酶标仪在540 nm处测定溶液吸光度。实验结果表明,以PBS作为阴性对照时,血红细胞膜未受到破坏,溶液为澄清透明。当以去离子水为阳性对照时,由于细胞内渗透压变大,细胞膜被破坏,血红蛋白分子溢出,溶液变成红色。经过酶标仪测定,碳点浓度在0.2 ~2.0mg/mL范围内,溶血率均低于国家药典规定的溶血度不超过5%的标准,即碳点具有良好的生物安全性。
将2×104个MCF-7细胞接种在15 mm内径的激光共聚焦专用培养皿中,并在含10% FBS(V/V)的 RPMI-1640 培养基中过夜培养,使其贴壁生长。加入0.5 mg/mL碳点溶液,并在 37℃继续孵育 4 h。孵育结束后,加入一定量 4%多聚甲醛在 4 ℃孵育 10 min,并用 PBS 清洗 3 遍。细胞固定后,用 PBS 清洗 3 遍。调节激光共聚焦的激发波长,对碳点在MCF-7细胞内拍照。图23为细胞MCF-7明场照片。当激发波长为405 nm激发时,碳点在细胞内呈蓝色荧光,且大量碳点在细胞质中(图24)。分别以458 nm和514 nm激发时,碳点在细胞内呈绿色荧光(图25)和红色荧光(图26)。
荧光碳点对日落黄检测:
配制含有不同浓度日落黄的0.1 mg/mL碳点溶液,测定315-580 nm波长范围内发射光谱。随着日落黄浓度提高,碳点溶液的强度降低(图27)。日落黄浓度在10~50μM,碳点的荧光强度具有线性变化,方程为:y=0.9486x+6.0695,根据检测限公式:LOD=3σ/S,计算出碳点对日落黄含量检测限为0.098μM(图28)。
- 还没有人留言评论。精彩留言会获得点赞!