一种稀土掺杂碱土金属硫化物纳米材料的制备方法与流程
本发明属于纳米发光材料技术领域,具体涉及一种稀土掺杂碱土金属硫化物纳米材料的制备方法。
背景技术:
近年来,稀土掺杂无机纳米发光材料得到广泛关注,由于其优异的性能,这些材料广泛应用在三维立体显示、防伪技术、固态激光器、光存储、太阳能电池、生物检测以及成像等方面。目前关于稀土掺杂无机纳米荧光材料的研究主要集中在稀土掺杂氟化物(如nayf4,nagdf4,caf2)等体系,对稀土掺杂碱土金属硫化物(ms)(m=ca,sr,ba)纳米发光材料的研究甚少。
稀土掺杂碱土金属硫化物具有立方相结构,空间点群为fm-3m,其八面体晶体场环境及其s2-的高共价性使得其具有不同于氟化物纳米发光材料优异的发光性能,如长余辉,光激励等。同时其在阴极射线、x射线、激光,可见光等激发下可实现高效可调谐荧光发射,因而被广泛地应用于led、光存储以及投影电视等方面。同时,这些性质也表明稀土掺杂碱土金属硫化物在生物成像和生物荧光标记等方面的应用潜力。
目前形貌均一、分散性好且尺寸可控的稀土掺杂碱土金属硫化物纳米材料的控制合成仍是该领域的一个技术挑战。其合成主要是采用前驱体在高温溶剂中发生热分解的方法。其中,meijerink等报道了采用二异丙基二硫代氨基甲酸金属盐作为前驱体进行合成,所合成的纳米颗粒为近球形,在10nm左右,分散性较好,但该合成方法所用前驱体二异丙基二硫代氨基甲酸金属盐无法购买,只能自己合成,且合成步骤繁琐,反应耗时久,纯度不易控制,都将影响纳米材料的合成,同时该合成方法未能实现纳米材料的尺寸调控(andriesmeijerinketal."lanthanide-dopedcasandsrsluminescentnanocrystals:asingle-sourceprecursorapproachfordoping",j.am.chem.soc.2014,136,16533-16543)。
技术实现要素:
为了改善现有技术的不足,本发明提供一种稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln的制备方法,ms:xmol%ln中m选自碱土金属ca,sr或ba;s代表硫元素;ln代表镧系稀土离子;xmol%代表镧系稀土离子占碱土金属与镧系稀土离子摩尔总数的百分比,0<x≤30mol%;所述制备方法包括如下步骤:
1)将碱土金属ca,sr或ba的盐,镧系稀土盐溶于溶剂r中,形成盐溶液;
2)向步骤1)得到的溶液中加入n’n-二苯基硫脲(dptu)的溶液;
3)将步骤2)得到的溶液进行加热,得到所述稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln;
溶剂r选自油酸、油胺及三辛胺和/或十八烯的混合物;
步骤2)中n’n-二苯基硫脲(dptu)的溶液使用的溶剂选自醇类溶剂、醚类溶剂、含氮的有机溶剂、酮类溶剂、卤代烷烃类溶剂或芳烃类溶剂。
根据本发明的制备方法,ms:xmol%ln中ln选自ho、er、tm、tb、eu、ce、sm、dy、nd、pr、gd、yb中的至少一种;所述ln的价态为3+或2+。
根据本发明的制备方法,ms:xmol%ln中0.05%≤x≤25mol%,优选为0.07%≤x≤22mol%。
根据本发明的制备方法,所述稀土掺杂碱土金属硫化物纳米材料为纳米晶ms:xmol%ln,其粒径为5~100nm,优选为10~50nm。
根据本发明的制备方法,步骤1)中,所述碱土金属ca,sr或ba的盐选自ca,sr或ba的卤盐、ca,sr或ba的醋酸盐、ca,sr或ba的硝酸盐、或三种盐的结晶水合物;优选为醋酸钙或其结晶水合物、醋酸锶或其结晶水合物、醋酸钡或其结晶水合物。
根据本发明的制备方法,步骤1)中,镧系稀土盐选自ho、er、tm、tb、eu、ce、sm、dy、nd、pr、gd、yb的醋酸盐、醋酸盐的结晶水合物中的至少一种。
根据本发明的制备方法,步骤1)中,溶剂r中油酸、油胺、三辛胺和/或十八烯的体积比为(1-10):(1-10):(1-100),优选为(1-6):(1-6):(1-50),还优选为(1-2):(1-6):(2-6),例如为1:2:2,2:3:5,1:3:6或1:6:3。
根据本发明的制备方法,步骤1)中,碱土金属ca,sr或ba的盐,镧系稀土盐的摩尔比为(1-x):x。
根据本发明的制备方法,步骤2)中n’n-二苯基硫脲(dptu)的溶液使用的溶剂选自乙醇、乙醚、二甲基甲酰胺、丙酮、氯仿或苯。
根据本发明的制备方法,步骤2)中,所述n’n-二苯基硫脲(dptu)的用量为碱土金属ca,sr或ba的盐与镧系稀土盐摩尔数之和的1~5倍,优选为2~4倍,还优选为3倍。
根据本发明的制备方法,步骤3)中,反应的温度为80-400℃,优选为80-330℃。
根据本发明的制备方法,步骤1)~3)优选在惰性气体氛围下进行,例如氮气氛围下进行。
优选地,所述制备方法还包括步骤4),步骤4)为将步骤3)制备的稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln与磷脂-聚乙二醇-氨基(dspe-peg-nh2)的氯仿溶液混合,得到水溶性的ms:xmol%ln纳米材料。
根据本发明的制备方法,步骤4)中所述dspe-peg-nh2与ms:xmol%ln的摩尔比为(1:1)-(100:1),优选为(1:1)-(50:1)。
根据本发明的制备方法,所述稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln可以为单层核壳结构ms:xmol%ln@ms,其中m、s、xmol%、ln具有上文所述的定义;当其为单层核壳结构时,所述制备方法还包括如下步骤:
a1)将步骤3)制备的稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln作为内核籽晶与碱土金属ca,sr或ba的盐、n’n-二苯基硫脲(dptu)的溶液混合后加热得到单层核壳结构ms:xmol%ln@ms;
任选地,将得到单层核壳结构ms:xmol%ln@ms与磷脂-聚乙二醇-氨基(dspe-peg-nh2)的氯仿溶液混合,得到水溶性的纳米材料。
根据本发明的制备方法,步骤a1)中稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln、碱土金属ca,sr或ba的盐、n’n-二苯基硫脲(dptu)的摩尔比为1:(1~3):(1~3)。
根据本发明的制备方法,步骤a1)中,反应的温度为80-400℃,优选为80-330℃。
根据本发明的制备方法,步骤a1)优选在惰性气体氛围下进行,例如氮气氛围下进行。
根据本发明的制备方法,所述稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln还可以为多层核壳结构ms:xmol%ln(@ms)n,其中m、s、xmol%、ln具有上文所述的定义;n为2以上的整数;当其为多层核壳结构时,所述制备方法还包括如下步骤:
b1)将步骤a1)制备的单层核壳结构ms:xmol%ln@ms作为内核籽晶与碱土金属ca,sr或ba的盐、n’n-二苯基硫脲(dptu)的溶液混合后加热;
b2)重复步骤b1),得到2层以上的多层核壳结构ms:xmol%ln(@ms)n;
任选地,将得到的多层核壳结构ms:xmol%ln(@ms)n与磷脂-聚乙二醇-氨基(dspe-peg-nh2)的氯仿溶液混合,得到水溶性的纳米材料。
根据本发明的制备方法,n选自2、3、4、5、6、7、8、9或10。
根据本发明的制备方法,步骤b1)中稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln、碱土金属ca,sr或ba的盐、n’n-二苯基硫脲(dptu)的摩尔比为1:(1~3):(1~3)。
根据本发明的制备方法,步骤b1)中,反应的温度为80~400℃,优选为80~330℃。
根据本发明的制备方法,步骤b1)优选在惰性气体氛围下进行,例如氮气氛围下进行。
本发明还提供如上所述方法制备得到的稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln。
本发明还提供如上所述方法制备得到的稀土掺杂碱土金属硫化物纳米材料ms:xmol%ln在光学存储、长余辉发光、照明显示、荧光标记检测、生物成像、光学防伪和编码中的用途。优选地,所述纳米材料用于荧光标记检测及生物成像。
作为实例,所述稀土掺杂碱土金属硫化物纳米材料可以选自如下材料:cas:0.07mol%ce3+纳米颗粒、cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒、cas:0.07mol%ce3+@cas@cas多壳层核壳结构纳米颗粒、srs:0.2mol%ce3+纳米颗粒、cas:0.07mol%ce3+,0.5mol%er3+纳米颗粒、cas:0.07mol%ce3+,0.2mol%nd3+纳米颗粒、cas:0.06mol%eu2+纳米颗粒、cas:0.06mol%eu2+,0.1mol%sm3+纳米颗粒、cas:20mol%yb3+,2mol%er3+纳米颗粒。
有益效果
1.本发明提供了一种稀土碱土金属硫化物纳米材料的制备方法,所述方法合成条件易于控制,反应原料无需自己制备,可以直接购买,节约成本和时间,反应周期短,过程简单,重复性好,原子利用率高,易于放大合成。
2.本发明的稀土掺杂碱土金属硫化物纳米晶在制备过程中可在80至400℃的宽温度区间合成目标产物,通过改变原料配比、溶剂配比、反应温度和/或反应时间可以有效地调控纳米材料的尺寸。
3.本发明的稀土掺杂碱土金属硫化物纳米材料稳定性好、纳米颗粒尺寸形貌均一,分散性好,荧光量子产率高、可实现全谱段(480-1600nm)发光,以实现其在生物检测以及生物成像方面的应用。
4.本发明的方法可以根据使用需要制备得到水溶性或油溶性的材料,所得材料为纯立方相结构,尺寸形貌均一,发光性能优异。
附图说明
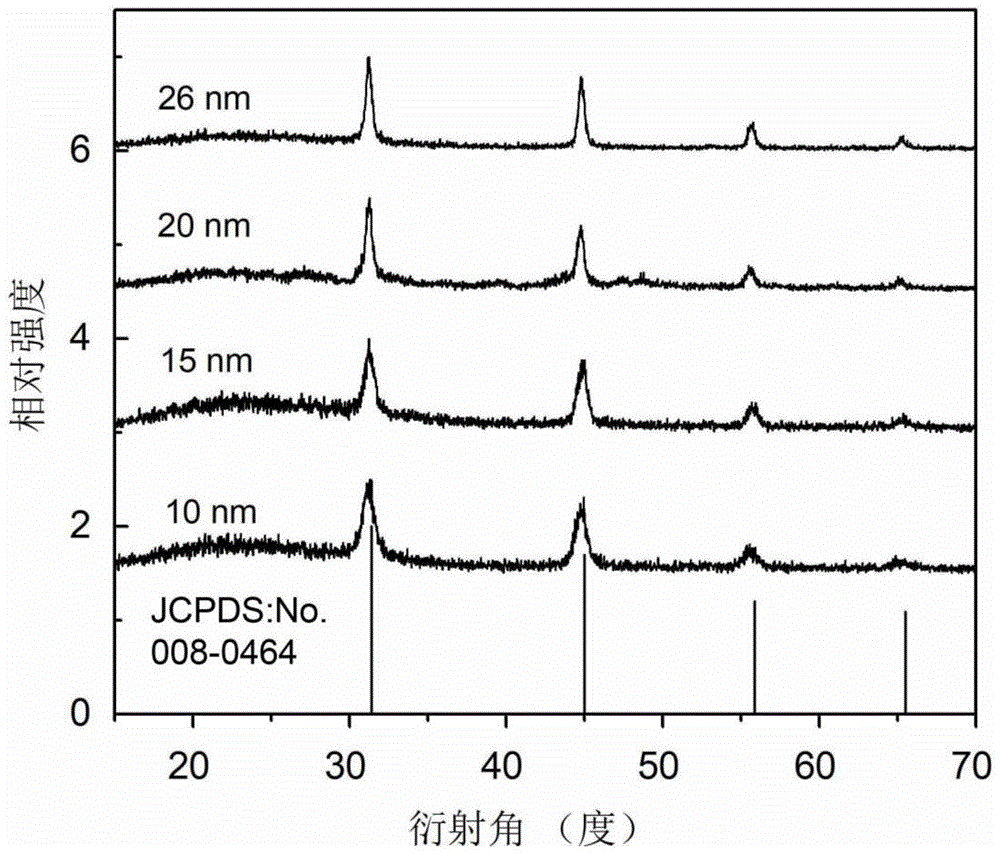
图1为实施例1中不同尺寸cas:0.07mol%ce3+纳米颗粒的粉末衍射图(其中jcpds:no.008-0464为cas纳米颗粒的标准pdf卡片号)。
图2为实施例1中不同尺寸cas:0.07mol%ce3+纳米颗粒的透射电镜图及其相应的粒径统计图。
图3为实施例2制备得到的cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒和实施例3制备得到的cas:0.07mol%ce3+@cas@cas多层核壳结构纳米颗粒的粉末衍射图(其中jcpds:no.008-0464为cas纳米颗粒的标准pdf卡片号)。
图4中(a)为实施例2制备得到的cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒和(b)实施例3得到的cas:0.07mol%ce3+@cas@cas多层核壳结构纳米颗粒的透射电镜图以及两者的电子衍射图。
图5为实施例4制备得到的srs:0.2mol%ce3+纳米颗粒的粉末衍射图、透射电镜图,及其x射线能谱分析图(其中jcpds:no.008-0489为srs纳米颗粒的标准pdf卡片号)。
图6为实施例1中不同尺寸cas:0.07mol%ce3+纳米颗粒的发射光谱图(激发450nm)(从上到下依次为粒径为26nm、20nm、15nm、10nm材料的检测结果)。
图7为实施例4制备得到的srs:0.2mol%ce3+纳米颗粒的发射光谱图(激发440nm)。
图8为实施例5制备的cas:0.07mol%ce3+,0.5mol%er3+纳米颗粒和实施例6制备的cas:0.07mol%ce3+,0.2mol%nd3+纳米颗粒的发射光谱图。
图9为实施例7制备的cas:0.06mol%eu2+纳米颗粒和实施例8制备的cas:0.06mol%eu2+,sm3+纳米颗粒的发射光谱图、余辉衰减图(激发470nm)以及光激励发射图(紫外灯辐照,980nm激光器激发)。
图10为实施例9制备的cas:20mol%yb3+,2mol%er3+纳米颗粒的制备的粉末衍射图和透射电镜图以及x射线能谱分析图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。此外,应理解,在阅读了本发明所公开的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本发明所限定的保护范围之内。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
仪器和设备:
本发明实施例产品进行粉末衍射表征使用的仪器型号为miniflex2,厂家为rigaku,铜靶辐射波长为λ=0.154187nm。
本发明实施例产品进行x射线能谱分析使用的仪器型号为jem-2010,厂家为jeol。
本发明实施例产品进行透射电镜检测使用的仪器型号为tecnaig2f20,厂家为fei。
本发明实施例产品进行紫外可见吸收光谱表征使用的仪器型号为lambda365,厂家为perkin-elmer。
本发明实施例产品进行荧光发射光谱、荧光寿命表征使用的仪器型号为fls980,厂家为edinburgh,激发光源为氙灯和450nm脉冲激光器。
实施例1:不同尺寸cas:0.07mol%ce3+纳米颗粒的制备
称取0.9993mmolca(ch3coo)2·h2o、0.0007mmolce(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.07mol%ce3+纳米颗粒;同时,在其他反应条件不变的情况下,改变溶剂油酸、油胺、三辛胺的体积比为2:3:5,1:3:6,1:6:3可以对应合成粒径为10nm,20nm,26nm的油溶性的cas:0.07mol%ce3+纳米颗粒。
实施例2:cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒的制备
称取0.9993mmolca(ch3coo)2·h2o、0.0007mmolce(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.07mol%ce3+纳米颗粒。
将油溶性cas:0.07mol%ce3+纳米颗粒作为内核籽晶,进行核壳包覆。称取1mmolca(ch3coo)2·h2o然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液c,然后降至室温;加入1mmol上述cas:0.07mol%ce3+内核籽晶。搅拌10min使之充分混匀,称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液c中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液d;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到25nm左右的油溶性cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒。
实施例3:cas:0.07mol%ce3+@cas@cas多壳层核壳结构纳米颗粒的制备
称取0.9993mmolca(ch3coo)2·h2o、0.0007mmolce(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.07mol%ce3+纳米颗粒。
将油溶性cas:0.07mol%ce3+纳米颗粒作为内核籽晶,进行核壳包覆。称取1mmolca(ch3coo)2·h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液c,然后降至室温;加入1mmol上述cas:0.07mol%ce3+内核籽晶。继续搅拌10min使之充分混匀,称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液c中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液d;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到25nm左右的油溶性cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒。
将上述油溶性cas:0.07mol%ce3+@cas单层核壳结构纳米颗粒作为内核籽晶,进行核壳包覆。称取1mmolca(ch3coo)2·h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液e,然后降至室温;加入1mmol上述cas:0.07mol%ce3+@cas内核籽晶。继续搅拌10min使之充分混匀,称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液f中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液g;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到30nm左右的油溶性cas:0.07mol%ce3+@cas@cas多壳层核壳结构纳米颗粒。
实施例4:srs:0.2mol%ce3+纳米颗粒的制备
称取0.998mmolsr(ch3coo)2·h2o、0.002mmolce(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到12nm左右的油溶性srs:0.2mol%ce3+纳米颗粒。
实施例5:cas:0.07mol%ce3+,0.5mol%er3+纳米颗粒的制备
称取0.9943mmolca(ch3coo)2·h2o、0.0007mmolce(ch3coo)3·4h2o,0.005mmoler(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.07mol%ce3+,0.5mol%er3+纳米颗粒。
实施例6:cas:0.07mol%ce3+,0.2mol%nd3+纳米颗粒的制备
称取0.9973mmolca(ch3coo)2·h2o、0.0007mmolce(ch3coo)3·4h2o,0.002mmolnd(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.07mol%ce3+,0.2mol%nd3+纳米颗粒。
实施例7:cas:0.06mol%eu2+纳米颗粒的制备
称取0.9994mmolca(ch3coo)2·h2o、0.0006mmoleu(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.06mol%eu2+纳米颗粒。
实施例8:cas:0.06mol%eu2+,0.1mol%sm3+纳米颗粒的制备
称取0.9983mmolca(ch3coo)2·h2o、0.0.0006mmoleu(ch3coo)3·4h2o,0.001mmolsm(ch3coo)3·4h2o,然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:0.06mol%eu2+,0.1mol%sm3+纳米颗粒。
实施例9:cas:20mol%yb3+,2mol%er3+纳米颗粒的制备
称取0.78mmolca(ch3coo)2·h2o、0.2mmolyb(ch3coo)3·4h2o,0.02mmoler(ch3coo)3·4h2o然后加入4ml油酸和8ml油胺和8ml三辛胺,通氮气加热至120℃并保温30分钟,形成透明溶液a,然后降至室温;称取3mmol的dptu,溶于10ml乙醇中,并逐滴加入到溶液a中,继续搅拌10min使之充分混匀,升温至80℃,保温30min,形成透明溶液b;升温至320℃,保温1h,降至室温;加入20ml乙醇沉淀分离并洗涤数次,得到15nm左右的油溶性cas:20mol%yb3+,2mol%er3+纳米颗粒。
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!