铑催化剂的制备方法与流程
1.本发明涉及化学催化领域,特别是涉及一种铑催化剂的制备方法。
背景技术:
2.催化对于化学化工来说具有重要意义,大多数的化学反应需要催化剂的参与,而其中需要贵金属参与的化学反应占到一半以上。尤其对于一些高选择性、需定向、低温低压进行的反应,贵金属的参与是很难替代的。然而铑非常昂贵,地壳中含量极少,铑催化剂的成本非常高。铑催化剂作为重要的催化材料,广泛应用于石油化工、医药化工、精细化工和环保等领域,特别是不对称催化领域,其可在手性配体作用下,广泛用于众多化工医药中间体的合成;例如不对称2,3
‑
二氢苯并[b]噻吩及其衍生物、苯基哌啶基吲哚衍生物以及α,β
‑
不饱和酯的不对称共轭1,4
‑
加成。
[0003]
以二(乙烯)氯铑(i)二聚体为例,目前主要由两种制备方法。一种方法:是将水合三氯化铑溶于甲醇或乙醇,再通入乙烯得到橙色粉末状产品,母液中加入氢氧化钠调节酸度。该方法会产生甲醛或乙醛等有毒气体的问题,且在实际生产中放大,由于该方法反应两次,操作较繁琐,反应效率低,致使部分氯化铑被还原为铑单质,大大降低了产品的收率和纯度,限制规模经营。另一种方法是:在高压釜中,先将三氯化铑水合物加入一定量的水中溶解,用高压乙烯和碱液加热搅拌反应制得产品。该方法涉及高压反应条件,对设备要求较高,且反应存在氯离子,氯离子在高压条件下容易腐蚀反应釜,该工艺的危险系数比较高。
技术实现要素:
[0004]
基于此,有必要提供一种工艺简单、无需高温高压,易于分离,能够提高收率和纯度的铑催化剂的制备方法。
[0005]
一种铑催化剂的制备方法,包括如下步骤:在三价铑盐的水溶液中通入乙烯或加入二烯烃,并于保护性气氛下加入锌粉进行还原反应,得到所述铑催化剂,所述铑催化剂为一价铑乙烯二聚体或一价铑二烯烃二聚体。
[0006]
在其中一些实施例中,在所述还原反应中,所述锌粉与所述三价铑盐的物质的量之比为(1~1.5):1。
[0007]
在其中一些实施例中,所述还原反应的条件为:于20~50℃下反应;和/或所述还原反应在常压下进行。
[0008]
在其中一些实施例中,所述还原反应具体包括如下步骤:于搅拌条件下分批加入所述锌粉,控制所述锌粉在反应体系的ph值为3.3~4.1时加完,继续反应至反应液的颜色逐渐褪去,并在1~2分钟内保持不变,控制所述还原反应结束。
[0009]
在其中一些实施例中,所述三价铑盐为三氯化铑;和/或所述二烯烃选自1,5
‑
环辛二烯及降冰片二烯中的至少一种。
[0010]
在其中一些实施例中,所述乙烯或所述二烯烃相对于所述三价铑盐是过量的。
[0011]
在其中一些实施例中,所述乙烯的通入速率为0.1l/min~0.5l/min;或者所述二烯烃与所述三价铑盐的物质的量之比为(1.1~2.2):1。
[0012]
在其中一些实施例中,在所述还原反应结束后,收集所述还原反应的沉淀,制得的铑催化剂为氯铑(i)乙烯二聚体或氯铑(i)二烯烃二聚体。
[0013]
在其中一些实施例中,还包括如下步骤:在所述还原反应之后,在所述还原反应的反应液中加入有机溶剂溶解沉淀,分液取有机相,于
‑
5℃~5℃加入乙酰丙酮金属盐或四氟硼酸盐进行离子交换反应。
[0014]
在其中一些实施例中,所述乙酰丙酮金属盐为乙酰丙酮钠;或者所述四氟硼酸盐为四氟硼酸钠和四氟硼酸银中的至少一种。
[0015]
上述铑催化剂的制备方法,采用锌粉在保护性气氛下还原三价铑盐,一锅法制得一价铑乙烯二聚体或一价铑二烯烃二聚体的铑催化剂,大大缩短反应时间,避免了多步反应操作繁琐降低产率的问题,此外采用锌粉作为还原剂,减少了有毒废气的产生,避免了采用甲醇或乙醇做还原剂产生甲醛或乙醛等有毒气体和采用高压乙烯和碱液加热搅拌反应对于设备要求高的问题。该制备方法采用一锅法,工艺简单,对设备要求低,无需高温高压,易于分离,适合放大生产,能够提高产物的收率和纯度。
附图说明
[0016]
图1为实施例7制得的产物的氢谱图;图2为实施例8制得的产物的氢谱图;图3为实施例9制得的产物的氢谱图;图4为实施例10制得的产物的氢谱图;图5为实施例11制得的产物的氢谱图。
具体实施方式
[0017]
为了便于理解本发明,下面将对本发明进行更全面的描述,并给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
[0018]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0019]
本发明一实施方式提供了一种铑催化剂的制备方法,包括如下步骤:在三价铑盐的水溶液中通入乙烯或加入二烯烃,并于保护性气氛下加入锌粉进行还原反应,得到铑催化剂,铑催化剂为一价铑乙烯二聚体或一价铑二烯烃二聚体。
[0020]
在上述还原反应的过程中锌粉逐渐溶解,三价铑被还原成一价铑,同时与乙烯或二烯烃配位生成不溶于水的铑催化剂,进而从反应液中析出,而氯化锌等副产物溶于水,易于分离,产物的收率较高。
[0021]
上述铑催化剂的制备方法,采用锌粉在保护性气氛下还原三价铑盐,一锅法制得一价铑乙烯二聚体或一价铑二烯烃二聚体的铑催化剂,大大缩短反应时间,避免了多步反
应操作繁琐降低产率的问题,此外采用锌粉作为还原剂,减少了有毒废气的产生,避免了采用甲醇或乙醇做还原剂产生甲醛或乙醛等有毒气体和采用高压乙烯和碱液加热搅拌反应对于设备要求高的问题。该制备方法采用一锅法,工艺简单,对设备要求低,无需高温高压,易于分离,适合放大生产,能够提高产物的收率和纯度。
[0022]
在其中一些实施例中,在还原反应中,锌粉与三价铑盐的物质的量之比为(1~1.5):1。通过优化锌粉与三价铑盐的物质的量之比,使得锌粉定量还原反应,以实现三价铑向一价铑的高效转化。
[0023]
在其中一些实施例中,还原反应的条件为:于20~50℃下反应。
[0024]
进一步地,还原反应的条件为:于20℃~35℃下反应。本发明铑催化剂的制备方法,采用锌粉在保护性气氛下还原三价铑盐,可优选在常温下进行,有利于促进向一价铑目标产物转化。
[0025]
进一步地,还原反应在常压下进行,其无需控制在高温高压下进行,故而无需在反应釜中进行,对设备要求低。实际操作中只要控制在保护性气氛下进行即可。进一步地,保护性气氛可为氩气、氮气或惰性气体等。
[0026]
在其中一些实施例中,还原反应具体包括如下步骤:于搅拌条件下分批加入锌粉,控制锌粉在反应体系的ph值为3.3~4.1时加完,继续反应至反应液的颜色逐渐褪去,并在1~2分钟内保持不变,控制还原反应结束。通过ph值和反应液颜色的监控,准确判断还原反应的终点,避免还原反应过度产生副产物铑单质,如此进一步提高了产物的收率。
[0027]
具体地,锌粉加入后产生橙红色沉淀,继续加入锌粉,控制锌粉在反应体系的ph值为3.3~4.1时加完,继续反应至反应液的颜色逐渐褪去,并在1~2分钟内保持不变,使得全部转化为橙红色沉淀。
[0028]
在其中一些实施例中,三价铑盐为三氯化铑,三价铑盐的选择不限于此,还可为其他可水溶的三价铑盐。
[0029]
在其中一些实施例中,二烯烃包括但不限于1,5
‑
环辛二烯(英文简称,cod)及降冰片二烯(英文简称,nbd)中的至少一种。
[0030]
在其中一些实施例中,乙烯或二烯烃相对于三价铑盐是过量的。
[0031]
进一步地,乙烯的通入速率为0.1l/min~0.5l/min;例如0.5l/min。
[0032]
进一步地,二烯烃与三价铑盐的物质的量之比为(1.1~2.2):1。
[0033]
更进一步地,铑催化剂中二烯烃与铑元素的物质的量之比为1:1,则优选二烯烃与三价铑盐的物质的量之比为(1.1~1.2):1。铑催化剂中二烯烃与铑元素的物质的量之比为2:1,则优选二烯烃与三价铑盐的物质的量之比为(2.1~2.2):1。
[0034]
在其中一些实施例中,在还原反应结束后,收集还原反应的沉淀,制得的铑催化剂为氯铑(i)乙烯二聚体或氯铑(i)二烯烃二聚体。
[0035]
在其中一些实施例中,还包括如下步骤:在还原反应之后,在还原反应的反应液中加入有机溶剂溶解沉淀,分液取有机相,于
‑
5℃~5℃加入乙酰丙酮金属盐或四氟硼酸盐进行离子交换反应。该离子交换反应的温度较为容易实现。
[0036]
通过上述离子交换反应,采用乙酰丙酮酰基团或四氟硼酸根离子替换中间体中氯离子等阴离子。该离子交换反应无需将还原反应的沉淀分离出来,可避免中间体转移造成产品损失,进而可提高离子交换后的产物的收率。
[0037]
进一步地,有机溶剂包括但不限于甲基叔丁基醚(mtbe)、乙醚和二氯甲烷中的至少一种。
[0038]
进一步地,在加入有机溶剂溶解沉淀后,保留有机相,并将水相继续用有机溶剂萃取,合并萃取的有机相和前述的有机相,在合并的有机相中加入乙酰丙酮金属盐或四氟硼酸盐进行离子交换反应。
[0039]
进一步地,在离子交换反应之后,还包括过滤取滤液,浓缩结晶,过滤干燥的步骤。在一具体示例中,浓缩结晶采用的溶剂是正己烷。
[0040]
进一步地,乙酰丙酮金属盐为乙酰丙酮钠,英文简写:na(acac)。进一步地,乙酰丙酮金属盐与对应的三价铑盐的物质的量之比为(1.0~1.5):1。
[0041]
进一步地,四氟硼酸盐为四氟硼酸钠和四氟硼酸银中的至少一种。进一步地,在加入四氟硼酸盐的同时,加入乙腈。在一具体示例中,四氟硼酸盐以其与乙腈的混合液的形式加入。试验证明,本发明的制备方法中在乙腈体系下,可采用四氟硼酸钠替换四氟硼酸银达到基本相同的技术效果,进而可避免四氟硼酸银存储条件苛刻、价格较为昂贵、重金属银离子的残留的问题,降低生产成本。
[0042]
进一步地,四氟硼酸盐与对应的三价铑盐的物质的量之比为(1.0~1.5):1。
[0043]
进一步地,四氟硼酸盐与乙腈的质量体积比为(4~10)g:200ml。
[0044]
为了更好地说明本发明,下面结合实施例对本发明内容作进一步说明。以下为具体实施例。
[0045]
二(乙烯)氯铑(i)二聚体的制备。即{rh(c2h4)2cl}2,mw:388.93,理论元素组成如下,c:24.71%;h:4.15%;rh:52.92%;结构式如下:实施例1:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温(本发明中是指25℃
±
5℃,下同)搅拌条件下分批加入2.4g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙红色沉淀,ph值为3.8时锌粉加完,反应液颜色逐渐褪去,ph值为4.1时在1
‑
2分钟内保持不变,停止通气。将沉淀无氧过滤,采用弱酸性的去离子水洗涤、干燥得到橙红色粉末6.3g。
[0046]
对比例1:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水100ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下滴加硼氢化钠0.7g和50ml去离子水的混合液; ph计在线检测ph值,ph值为1.1时反应液中开始出现黑色沉淀,随着硼氢化钠水溶液加完,反应液析出大量黑色沉淀推测为单质铑;没有将反应继续进行,此方案不可行。
[0047]
对比例2:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水100ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以
0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下滴加80%水合肼2.2g和20ml去离子水的混合液;ph计在线检测ph值,ph值为1.5时反应液中开始出现黑色沉淀,ph值为3.6时反应液颜色逐渐褪去,但是没有出现橙红色沉淀,沉淀全是黑色的,推测为单质铑,此方案不可行。
[0048]
对比例3:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水100ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下滴加抗坏血酸钠14.7g和50ml去离子水的混合液;ph计在线检测ph值,ph值为2.4时反应液中开始出现橙红色沉淀,柠檬酸三钠水溶液加完,反应液继续通气2天,ph值为4.8保持不变,停止通气。将沉淀无氧过滤,采用去离子水洗涤、干燥得到橙红色粉末1.4g。
[0049]
对比例4:500ml三口瓶加入三水合三氯化铑10.0g,去离子水50ml,室温条件下搅拌30分钟将三氯化铑完全溶解,加入甲醇240ml;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;6小时后,ph计在线检测ph值,ph值为1.4时反应液中开始出现橙红色沉淀,48小时后ph值为2.2时基本保持不变,反应液颜色依然为红色,停止通气。将沉淀无氧过滤,去离子水洗涤,滤液加1m氢氧化钠水溶液,调节ph3
‑
4,又析出少量得到橙红色粉末,无氧过滤,合并两次滤饼干燥得到橙红色粉末5.6g。
[0050]
实施例2:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.2时反应液中开始出现橙红色沉淀,ph值为3.8时锌粉加完,反应液颜色逐渐褪去,ph值为4.3时在1
‑
2分钟内保持不变,停止通气。将沉淀无氧过滤,采用弱酸性的去离子水洗涤、干燥得到橙红色粉末6.7g。
[0051]
实施例3:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下分批加入3.6 g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙红色沉淀,ph值为3.9时锌粉加完,反应液颜色逐渐褪去,ph值为4.5时在1
‑
2分钟内保持不变,停止通气。将沉淀无氧过滤,弱酸性的去离子水多次洗涤、干燥得到橙红色粉末6.2g。
[0052]
实施例4: 250ml三口瓶加入三水合三氯化铑10.0g,去离子水100ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;加热至反应体系的温度为50℃搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.2时反应液中开始出现橙红色沉淀,ph值为3.9时锌粉加完,反应液颜色逐渐褪去,ph值为4.1时在1
‑
2分钟内保持不变,停止通气。将沉淀无氧过滤,采用弱酸性的去离子水洗涤、干燥得到橙红色粉末5.8g。
[0053]
乙酰丙酮酰双(亚乙基)化铑(i)的制备。即rh(c2h4)2(acac);mw:258.13。理论元素组成如下,c:41.88%;h:5.86%;rh:39.87%;结构式如下:
实施例5:2l三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙红色沉淀,ph值为4.1时锌粉加完,反应液颜色逐渐褪去,ph值为4.4时在1
‑
2分钟内保持不变,停止通气。加入甲基叔丁基醚(mtbe)1.2l溶解沉淀,无氧分液,水相用甲基叔丁基醚萃取3次,合并滤液有机相冷却0℃,分批加入乙酰丙酮钠固体6.2g,加完在0℃搅拌1小时,无水无氧过滤,滤液浓缩,加入正己烷,无水无氧过滤干燥得到橙色粉末8.8g。
[0054]
实施例6:2l三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解;先用氩气置换气氛三次,再用乙烯气体置换三次,并以0.5l/分钟的通气速度向反应液中通入乙烯气体;常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙红色沉淀,ph值为4.1时锌粉加完,反应液颜色逐渐褪去,ph值为4.4时在1
‑
2分钟内保持不变,停止通气。加入甲基叔丁基醚1.2l溶解沉淀,无氧分液,水相用无水乙醚萃取3次,合并滤液有机相冷却0℃,分批加入乙酰丙酮钠固体6.2g,加完在0℃搅拌1小时,无水无氧过滤,滤液浓缩,加入正己烷,无水无氧过滤干燥得到橙色粉末8.7g。
[0055]
具体地,本发明中产物的收率的计算方式为产物中金属铑的理论重量/原料中金属铑的理论重量
×
100%。本发明中产物的纯度的计算方法为产物中铑含量检测值/产物中理论铑含量
×
100%。其中,原料三水合三氯化铑中金属铑含量为38wt%。
[0056]
上述各实施例和对比例的部分参数和性能测试结果如下表所示:
通过对比例1~2可以看出,硼氢化钠和水合肼不能合成目标产物,此方案不可行,可能是其还原性太强,使得三氯化铑被还原成单质铑。
[0057]
通过对比例3~4可以看出,抗坏血酸钠和甲醇的还原性较弱,反应时间比较长,收率偏低。且对比例4采用甲醇做还原剂,存在产生甲醛等有毒废弃的缺点。而本发明实施例采用锌粉的实施例的反应时间短,收率较高,且无废弃问题。
[0058]
通过实施例2~6和实施例1可以看出,实施例2中的锌粉当量在1.1时,收率最高。通过实施例4和实施例2可以看出,锌粉当量为1.1,且同时在加热条件下还原,存在还原过度,伴随有单质铑产生的问题;故而优选在常温下进行。通过实施例4~5和实施例1可以看出,采用该制备方法进一步制得乙酰丙酮酰双(亚乙基)化铑(i),仍保持在较高收率。
[0059]
在上述试验探索的基础上,进一步以二烯烃替代上述的乙烯进行了如下的试验探索。
[0060]
(1,5
‑
环辛二烯)氯铑(i)二聚体的制备。{rh(cod)cl}2:mw:493.08;结构如下:
实施例7:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解,加入1,5
‑
环辛二烯6.0g;先用氩气置换气氛三次,常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙黄色沉淀,ph值为3.4时锌粉加完,反应液颜色逐渐褪去,继续搅拌反应;ph值为4.2时在1
‑
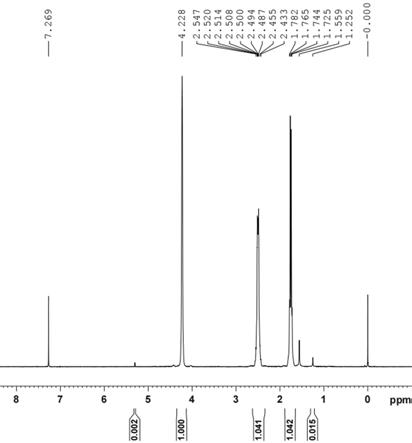
2分钟内保持不变停止搅拌,将沉淀空气中过滤,去离子水洗涤、干燥得到橙黄色粉末8.5g。计算产物的收率为94%。产物的1hnmr cdcl
3 见附图1。
[0061]
降冰片二烯氯化铑(i)二聚体的制备。即:{rh(nbd)cl}2,mw:461.00;结构式如下:实施例8:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解,加入降冰片二烯5.1g;先用氩气置换气氛三次,常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.0时反应液中开始出现橙黄色沉淀,ph值为3.3时锌粉加完,反应液颜色逐渐褪去,继续搅拌反应;ph值为4.2时在1
‑
2分钟内保持不变停止搅拌,将沉淀空气中过滤,去离子水洗涤、干燥得到土黄色粉末7.9g。计算产物的收率为93%。产物的1hnmr cdcl
3 见附图2。
[0062]
二(1,5
‑
环辛二烯)四氟硼酸铑(i)的制备。即rh(cod)2bf4,mw:406.07;结构式如下:实施例9:1l三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解,加入1,5
‑
环辛二烯10g;先用氩气置换气氛三次,常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙黄色沉淀,ph值为3.4时锌粉加完,反应液颜色逐渐褪去,继续搅拌反应;ph值为4.2时在1
‑
2分钟内保持不变停止搅拌,加入二氯甲烷500ml将沉淀溶解,无氧分液,水相用二氯甲烷萃取三次,合并滤液有机相加入四氟硼酸钠4.5g和乙腈200ml混合液,室温搅拌过夜,无水无氧过滤,滤液浓缩加入正己烷析晶,无水无氧过滤,干燥得到红棕色粉末14.4g。计算产物的收率为96%。产物的1hnmr cdcl
3 见附图3。
[0063]
实施例10:1l三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解,加入1,5
‑
环辛二烯10g;先用氩气置换气氛三次,常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.1时反应液中开始出现橙黄色沉淀,
ph值为3.4时锌粉加完,反应液颜色逐渐褪去,继续搅拌反应;ph值为4.2时在1
‑
2分钟内保持不变停止搅拌,加入二氯甲烷500ml将沉淀溶解,无氧分液,水相用二氯甲烷萃取三次,合并滤液有机相加入四氟硼酸银8.6g和乙腈200ml混合液,室温搅拌过夜,无水无氧过滤,滤液浓缩加入正己烷析晶,无水无氧过滤,干燥得到红棕色粉末14.6g。计算产物的收率为97%。产物的1hnmr cdcl
3 见附图4。
[0064]
双(降冰片二烯)四氟硼酸铑(i)的制备。即rh(nbd)2bf4,mw:373.99;结构式如下:实施例11:在250ml三口瓶加入三水合三氯化铑10.0g,去离子水150ml,室温条件下搅拌30分钟将三氯化铑完全溶解,加入降冰片二烯5.1g;先用氩气置换气氛三次,常温搅拌条件下分批加入2.7g锌粉;ph计在线检测ph值,ph值为2.0时反应液中开始出现橙黄色沉淀,ph值为3.3时锌粉加完,反应液颜色逐渐褪去,继续搅拌反应;ph值为4.2时在1
‑
2分钟内保持不变停止搅拌,加入二氯甲烷500ml将沉淀溶解,无氧分液,水相用二氯甲烷萃取三次,合并滤液有机相加入四氟硼酸钠4.5g和乙腈200ml混合液,室温搅拌过夜,无水无氧过滤,滤液浓缩加入甲基叔丁基醚析晶,无水无氧过滤,干燥得到红色粉末12.7g。计算产物的收率为92%。产物的1hnmr cdcl
3 见附图5。
[0065]
上述各实施例和对比例的部分参数和性能测试结果如下表所示:
通过实施例7~11和附图1~图5可以看出,本发明的制备方法同样可适用于一价铑二烯烃二聚体的制备。
[0066]
通过实施例9~10可以看出,四氟硼酸钠在乙腈体系下可以替换四氟硼酸银,而收率相当。
[0067]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0068]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准,说明书可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1