立达霉有关物质的测定方法与流程
本发明涉及农药技术领域,具体地说,是立达霉有关物质的测定方法。
背景技术:
高效液相色谱法(highperformanceliquidchromatography,hplc)是色谱法的一个重要分支,以液体为流动相,采用高压输液系统,将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱中,在柱内各成分被分离后,进入检测器进行检测,从而实现对试样的分析。高效液相色谱法已在化学、医学、工业、农学、商检、法检等领域得到广泛的应用。
中国专利文献cn105223278a公开了一种固相萃取液质联用法测定葡萄酒中甲霜灵、多菌灵的方法,包括先调节待测葡萄酒的ph值,用乙腈对酒样进行液液萃取,加入氯化钠待溶液盐析后用固相萃取柱萃取乙腈层,通过固相萃取柱洗脱,收集净化后的洗脱液,继而转入旋转蒸发仪浓缩,用氮气吹干浓缩液,通过乙腈水溶液回溶后,注入液质联用仪进行分析,获得葡萄酒中甲霜灵、多菌灵的含量的步骤。中国专利文献cn104535671a公开了一种茄子中甲霜灵残留的检测方法,包括以下几个步骤:(1)样品处理:将采摘后的茄子置于破碎机中破碎,得到茄子浆;(2)提取:称取茄子浆20g于锥形瓶中,加入100ml的乙酸乙酯和丙酮混合液,在温度为35-40℃的条件下振荡提取60-70min,抽滤得清液,浓缩至近干,氮吹至干,用2ml乙腈复溶备用;(3)小柱分离:用5ml乙酸乙酯洗涤hlb小柱,再用5ml丙酮洗涤小柱,上样,用10ml体积比为1:2的乙腈和水混合液淋洗小柱,最后用10ml的乙腈洗脱,收集洗脱液,旋蒸至1ml,氮气吹干,采用乙腈定容至2ml,待检测;(4)检测:采用hplc进行检测。但是关于本发明的立达霉有关物质的测定方法目前还未见报道。
技术实现要素:
本发明的目的是针对现有技术中的不足,提供一种立达霉有关物质的测定方法。
为实现上述目的,本发明采取的技术方案是:一种立达霉有关物质的测定方法,所述的测定方法包括:
(1)专属性试验,
(2)稳定性试验,
(3)最大的有关物质测定。
一种立达霉专属性试验方法,所述的试验方法包括:
(1)酸破坏试验:取样品加盐酸溶液,置热水浴中加热,取出后放冷,用氢氧化钠中和,加流动相稀释,然后进行hplc分析;
(2)碱破坏试验:取样品加氢氧化钠,置热水浴中加热,取出后放冷,用盐酸中和,加流动相稀释,然后进行hplc分析;
(3)氧化破坏试验:取样品加过氧化氢,放置一段时间后取出,加流动相稀释,然后进行hplc分析;
(4)加热破坏试验:取样品加流动相稀释,置100℃水浴中加热,取出后放至室温,然后进行hplc分析;
(5)光破坏试验:取样品置于紫外灯下照射,加流动相稀释,然后进行hplc分析。
所述的试验方法包括:
(1)酸破坏试验:取样品20mg加1mol/l盐酸溶液5ml,置热水浴中加热30min,取出后放冷,用1mol/l氢氧化钠溶液中和,加流动相稀释制成每1ml中含2mg样品的溶液,然后进行hplc分析;
(2)碱破坏试验:取样品20mg加1mol/l氢氧化钠溶液5ml,置热水浴中加热30min,取出后放冷,用盐酸中和,加流动相稀释制成每1ml中含2mg样品的溶液,然后进行hplc分析;
(3)氧化破坏试验:取样品20mg加30%过氧化氢5ml,放置1小时后取出,加流动相稀释制成每1ml中含2mg样品的溶液,然后进行hplc分析;
(4)加热破坏试验:取样品加流动相稀释制成每1ml中含2mg样品的溶液,置100℃水浴中加热2小时,取出后放至室温,然后进行hplc分析;
(5)光破坏试验:取样品置于紫外灯下照射24小时,称取20mg加流动相稀释制成每1ml中含2mg的溶液,然后进行hplc分析。
hplc分析的条件为:c18柱,柱温是30℃,检测波长225nm,流速1.5ml/min,流动相为甲醇:水=5:95。
一种立达霉稳定性试验方法,所述的稳定性试验方法包括以下步骤:称取样品并用流动相溶解,在0-24h内在hplc进样分析,记录色谱图,以主峰峰面积考察溶液的稳定性。
一种立达霉最大的有关物质的测定方法,所述的测定方法为运用hplc测定2,6-二甲苯胺,色谱柱为c18柱,流动相为乙腈:磷酸水=40:60,检测波长为225nm,检测温度为30℃,流速为1.25ml/min,进样量为10μl。
2,6-二甲苯胺的检出限为0.02μg/ml。
2,6-二甲苯胺的定量限为0.1μg/ml。
本发明优点在于:
1、本发明采用hplc对立达霉有关物质进行测定,具有高效、分离效能高,灵敏度高,分析速度快,准确度高等优点。
2、本发明提供了立达霉的专属性试验方法、稳定性试验方法和最大的有关物质的测定方法。
附图说明
附图1:未破坏的系统适应性溶液色谱图。
附图2:光破坏立达霉的色谱图。
附图3:酸破坏立达霉的色谱图。
附图4:碱破坏立达霉的色谱图。
附图5:氧化破坏立达霉的色谱图。
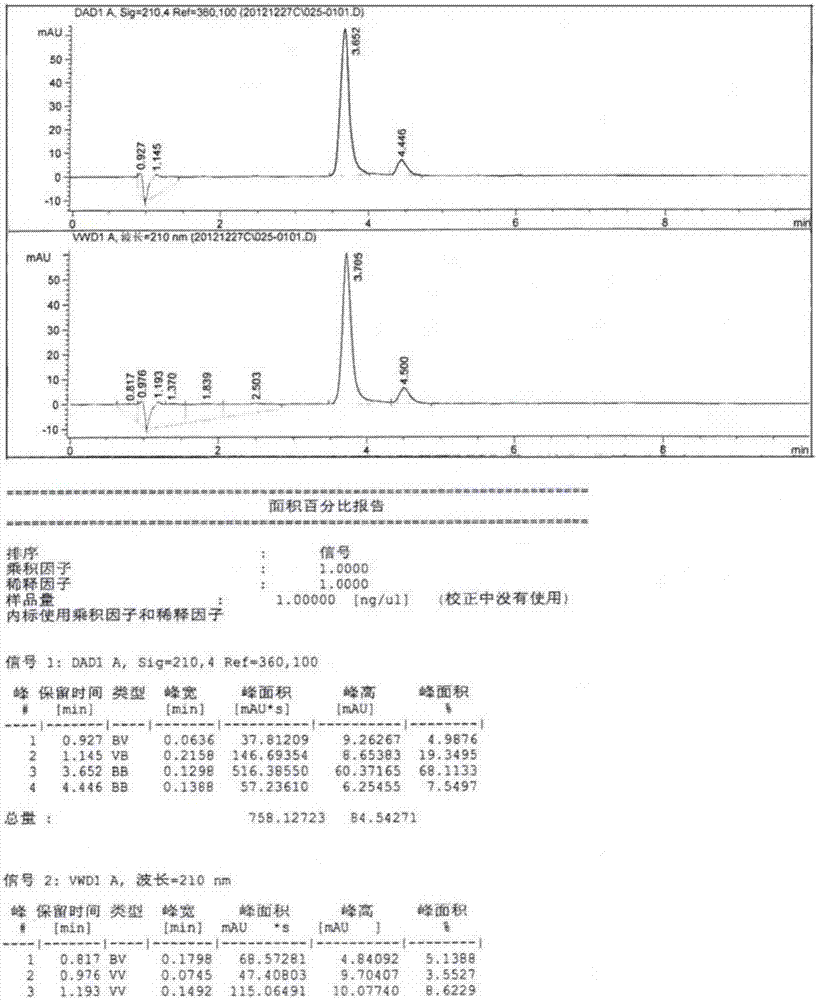
附图6:2,6-二甲苯胺的色谱图。
附图7。2,6-二甲苯胺和立达霉的色谱图。
附图8:0.1μg/ml的2,6-二甲苯胺的色谱图。
具体实施方式
下面对本发明提供的具体实施方式作详细说明。
对照品:
立达霉(甲霜灵)对照品:购自北京世纪奥科生物技术有限公司,德国dr.ehrenstorfer,cas编号57837-19-1(watercontent0.1%,det.purity99.0%,tolerance/uncertainty+/-0.5%,determinedbykarl-fischertitration)。
供试品:
“复方立达霉粉”(美婷),长沙拜特生物科技研究所有限公司提供,样品编号分别为20111101、20111201、20120101、20120508、20120708、20120722、20120811、20120813、20120830、20121101、20121201和20130101。
溶液制备
供试品溶液取本品适量,加流动相稀释至每1ml中约含立达霉0.1mg的溶液,作供试品溶液。
对照品溶液精密称取立达霉对照品适量,加流动相溶解并稀释为每1ml中约含0.1mg立达霉的溶液,作为对照品溶液。
1、专属性试验
酸破坏试验:取立达霉20mg,加1mol/l盐酸溶液5ml,置热水浴中,加热30min取出,放冷,用1mol/l氢氧化钠溶液中和,加流动相稀释制成每1ml中含2mg的溶液,上hplc。
碱破坏试验:取立达霉20mg,加1mol/l氢氧化钠5ml,置热水浴中,加热30min取出,放冷,用1mol/l盐酸溶液中和,加流动相稀释制成每1ml中含2mg的溶液,上hplc。
氧化破坏试验:取立达霉20mg,加30%过氧化氢5ml,放置1h,取出,加流动相稀释制成每1ml中含2mg的溶液,上hplc。
加热破坏试验:取立达霉适量,加流动相溶解并稀释成每1ml中含有2mg的溶液,置100℃水浴中,加热2h,取出,放至室温。上hplc。
光破坏试验:取立达霉适量,置紫外灯下,照射24h,称取20mg,精密测定,加流动相溶解并稀释成每1ml中含有2mg的溶液,上hplc。
根据制剂处方及工艺,制备空白辅料样品,上hplc。
取系统适用性溶液,上hplc。
hplc条件:色谱柱是rp-ods柱(150mm×4.6mm,5μm),柱温是30℃,检测波长包括225nm,流速1.5ml/min,流动相为甲醇:水=5:95(v:v)。
未破坏的系统适应性溶液色谱图见附图1,光破坏立达霉的色谱图见附图2,酸破坏立达霉色谱图见附图3,碱破坏立达霉色谱图见附图4,氧化破坏立达霉色谱图见附图5。
结果显示,立达霉经过酸/碱水浴、光和氧化破坏能产生明显的降解产物,立达霉在加热破坏时未出现降解产物。建立的hplc法能有效分离立达霉及其经酸碱、光和氧化破坏后产生的降解产物,立达霉峰与其它分解产物峰分离良好,说明本法专属性强。
2、稳定性试验
精密称取“复方立达霉粉”(批号20111101)适量,加流动相溶解,在0~24h内进样测定并记录色谱图,以主峰峰面积和杂质含量为指标考察溶液稳定性,结果见表1。立达霉主峰面积的rsd=0.25%。结果表明立达霉供试品溶液24h内稳定性良好。
表1稳定性试验结果
3、样品测定
将不同批次的“复方立达霉粉”按照以上方法(稳定性试验的操作方法)进行测定,结果见表2。在有关物质方面,“复方立达霉粉”样品中单个最大杂质最高为3.48%,最低为1.97%,平均为2.75%。“复方立达霉粉”样品中总杂质最高为5.32%,最低为3.24%,平均为4.48%。
表2样品有关物质测定结果
根据有关物质测定结果,将单个杂质限度定为4.0%,杂质总量限度定为6.0%。
4、最大的有关物质
最大的有关物质为2,6-二甲苯胺。2,6-二甲苯胺是立达霉合成途径过程的中间产物(详见《立达霉(甲霜灵)生产工艺》),有可能在生产立达霉原料过程中掺入到立达霉原料中。
2,6-二甲苯胺的分析方法采取高效液相色谱法,色谱条件为:c18rp-ods柱(150mm×4.6mm,5μm);流动相【乙腈:磷酸水=40:60(v/v)】;检测波长为225nm;检测温度30℃,流速为1.25ml/min。进样量10μl。该检测方法可以有效地将检测2,6-二甲苯胺,2,6-二甲苯胺保留时间约为1.68min(附图6)。将2,6-二甲苯胺和立达霉的混合液上hplc,2,6-二甲苯胺保留时间约为1.68min,立达霉的保留时间为4.97min(附图7),证明该方法特异性良好。
以信噪比s/n≥3计算,确定2,6-二甲苯胺的检出限为0.02μg/ml,相当于总含量的0.01%。以信噪比s/n≥10计算,确定2,6-二甲苯胺的定量限为0.1μg/ml(附图8),相当于总含量的0.05%。以上比例均远低于杂质的限度值。
将系列浓度梯度的2,6-二甲苯胺标准溶液上hplc。以2,6-二甲苯胺浓度(μg/ml)为横坐标,以峰面积为纵坐标绘制标准曲线。2,6-二甲苯胺标准溶液在0.1μg/ml-1000μg/ml的线性范围内与峰面积的线性关系良好,线性回归方程为y=5.5254x+6.1058,相关系数(r2)为0.9999。
将一定量2,6-二甲苯胺添加到空白溶液中,用以上方法,测定2,6-二甲苯胺的准确度和精密度。2,6-二甲苯胺的平均回收率为94.60%~99.02%,精密度为0.55%~4.45%,详细见表3。
表32,6-二甲苯检测的准确度和精密度(n=5)
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!