一种膜蛋白检测制剂、非标记肿瘤纳米探针及使用方法与流程
本发明属于膜蛋白检测技术领域;具体涉及一种基于膜蛋白能够引起c-rich的释放,通过银纳米簇从聚丙烯酸(paa)框架上转移到c-richdna序列上的非标记原位膜蛋白检测制剂、非标记肿瘤纳米探针及它们的使用方法。
背景技术:
膜蛋白是生物膜中所含有的蛋白,是生物膜的主要功能承担者,在生物生理功能中占有举足轻重的作用。由于膜蛋白特殊的功能性质,使得膜蛋白一旦发生变化,就会导致人类许多疾病的发生。例如,细胞膜上面的某些膜蛋白发生变化,就会导致正常细胞的生理功能发生紊乱,有可能会导致正常细胞转换成恶性肿瘤细胞。而由于膜蛋白在细胞表面,很容易与外界分子接触,因此在许多的生物工程中,膜蛋白被用来当作药物靶点,或者用于标记肿瘤细胞,也因此在肿瘤临床治疗上具有非常良好的应用前景。然而,许多的技术尽管能够进行敏感准确地完成膜蛋白检测,但是这些技术通常需要分离、净化等繁琐的预处理过程,以达到膜蛋白原位检测的目的。
目前,有许多利用核酸适配体技术进行原位检测膜蛋白的方法。核酸适配体通常是一小部分的dna、rna、xna或者肽,是一种抗体类似物,它能够与多种生物分子高特异性、高选择性地结合,而且其尺寸小,具有更好的组织穿透能力,因此被广泛用于生物传感器领域。但是,单纯的只是利用核酸适配体技术进行原位检测膜蛋白也有一些缺点,比如说目标膜蛋白需要具有荧光特性的荧光染料标记之后,而这种利用化学物质标记的方法需要精密的仪器和受过训练的专业人员人员进行操作检测,这就增大了膜蛋白检测的成本。
银纳米簇(agncs)由于具有很强的量子限制效应和小尺寸效应,表现出理想的光学和电学性能,从而成为纳米材料研究领域的热点之一。相较于有机染料荧光染料易漂白的性质,银纳米簇发光颜色随团簇尺寸可调,并且能够在非常温和的条件下就能合成,其作为一种潜在的荧光标记物,有望应用于光成像、生物标记、化学传感器等领域。由于agncs本身具有的量子限制效应,使得其与胞嘧啶能够稳定结合,agncs就能与原来的模板发生剥离,进而与富含胞嘧啶的单链(c-rich)dna序列结合。
技术实现要素:
为解决现有检测膜蛋白方法的步骤繁琐及造价高的问题,本发明提供了一种基于膜蛋白引发c-rich序列的释放,接着利用将agncs从原有模板转移到c-rich序列上,从而达到无需标记就能实现原位检测膜蛋白的制剂。旨在提高原位检测膜蛋白的灵敏度,降低实验成本,简化实验步骤。
一种膜蛋白检测制剂,包括:聚丙烯酸(paa)保护的agncs、由核酸适配子和富含胞嘧啶的单链(c-rich序列)杂合形成的双链dna。
所述的聚丙烯酸(paa)保护的agncs实际上是由聚丙烯酸与硝酸银溶液混合反应得到,加入适量的硼氢化钠还原溶液中的银离子,终产物中聚丙烯酸与agncs紧密结合,且用于检测时能提供足够的agncs与c-rich序列结合;反应完成后离心洗涤,透析以去除硼氢化钠和多余银离子。
作为进一步的优选,所述的膜蛋白是酪激酶-7(ptk7),这种膜蛋白通常被用作t细胞急性淋巴细胞白血病(t-all)细胞的标记物以及受体。而且本发明仅限于检测膜蛋白,因为如:bsa、has和fn蛋白均不能与ag特异性结合产生荧光。
只选c-rich序列与适配子杂合,而不是a-rich、g-rich和t-rich,源于ag与胞嘧啶的结合能力要强于其他三种脱氧核糖。
选用的c-rich序列中胞嘧啶的含量对于dna-agncs的荧光强度影响很大,原因是由于胞嘧啶中的n3能与ag原子的结合,一定程度下,含有数量愈多,结合能力愈大。
选用的c-rich序列中胞嘧啶的位置对于dna-agncs的荧光强度影响很大,一味的增大胞嘧啶数目并不能持续地增大荧光强度,原因在于,胞嘧啶的位置也即胞嘧啶之间的间隙也会对荧光强度产生很大影响。
作为进一步的优选,所述的富含胞嘧啶的单链的胞嘧啶数目占30%以上。
作为进一步的优选,所述的富含胞嘧啶的单链每3个胞嘧啶之间间隔至少一个其他的脱氧核糖。会增大荧光强度,原因是ag与胞嘧啶之间的位阻效应。
作为进一步的优选,所述的核酸适配子为sgc8,序列为:5’-atctaactgctgcgccgccgggaaaatactgtacggttaga-3’;富含胞嘧啶的单链为s16,序列为5’-tctaaccctccctcccgtacag-3’。
本发明的第二个目的是提供非标记肿瘤纳米探针,该探针即上述的由核酸适配子和富含胞嘧啶的单链(c-rich序列)杂合形成的双链dna。
本发明的第三个目的是提供所述的膜蛋白检测制剂的使用方法,选用聚丙烯酸保护的agncs、由核酸适配子和富含胞嘧啶的单链杂合形成的双链dna混合,其中适配子能够与膜蛋白强力结合,导致dna双螺旋构象的改变,从而导致富含胞嘧啶的单链释放,接着富含胞嘧啶的单链与聚丙烯酸框架上面的agncs强力结合,导致纳米粒子的荧光强度放大。
作为进一步的优选,膜蛋白检测体系中,ph取值为5.5-6.0,优选5.5;温度在40-50℃优选45℃;反应时间3.5-8分钟,优选4-6分钟;膜蛋白ptk7的检测范围从30pm-2nm,最小检测限为12pm。
ph能够影响agncs转运效率,ph低于4.5时,未发生agncs转移到dna反应,随着ph的增大,转移效率逐渐增大,直到ph增加到5.5,此时荧光强度最大,ph大于6时,agncs转运效率明显减小。
温度能够影响agncs转运效率,温度变化范围在25-45℃时,转移效率逐渐增大,荧光强度。温度再增加时,agncs转运效率逐渐减小。
作为进一步的优选,膜蛋白检测中,在加入dna之后的第4min开始选作传输时间(传输时间是指agncs与paa分离,进而与dna单链结合的时间)。在加入dna之后的第4min荧光强度迅速增加,因此可以选作最优传输时间。
作为进一步的优选,膜蛋白检测中,膜蛋白ptk7的检测范围从30pm-2nm,最小检测限为12pm。
本发明与其他依赖人工与仪器的复杂的检测方法相比,利用该方法能够灵敏快速准确的进行膜蛋白的检测,而且能够定量测量膜蛋白,这使得该方法在一些疾病的研究以及临床诊断上具有良好的应用前景。
附图说明
图1a和b分别为激发波长在470nm激发和550nm荧光发射光谱,实线曲线代表paa-agncs,点画线曲线代表dna-agncs。
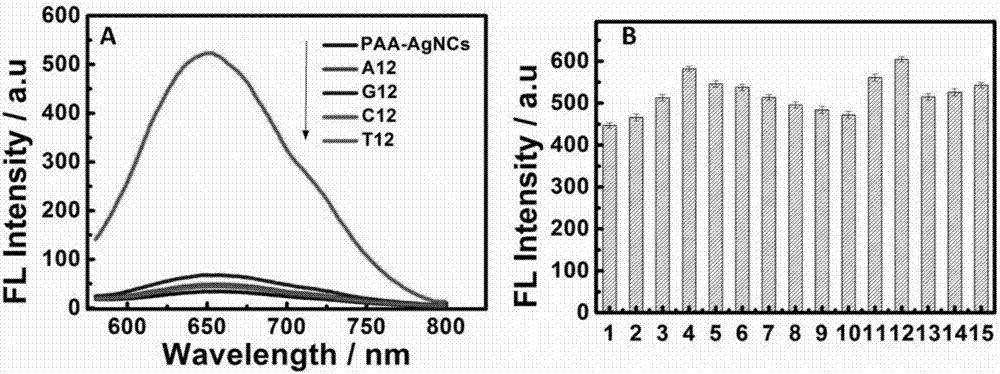
图2a黑色曲线代表paa-agncs,曲线分别代表c12与g12、a12、t12、paa-agncs结合后的荧光光谱图。b代表15组分别含有12个寡核苷酸,但由不同的胞嘧啶和胸腺嘧啶组合,不同数量及不同位点胞嘧啶下与agncs结合之后的荧光强度。
1、2、3、4、5、6、7、8、9、10、11、12、13、14、15柱状图分别代表s1、s2、s3、s4……s15与agncs结合之后的荧光强度,s1:5’-ttttcccctttt;s2:5’-tttccccccttt;s3:5’-ttcccccccctt;s4:5’-tcccccccccct;s5:5’-cccccccccccc;s6:5’-ttcctcctcctt;s7:5’-ttccttccttcc;s8:5’-cctttcctttcc;s9:5’-ccttttccttttcc;s10:5’-cctttttcctttttcc;s11:5’-tcctcctcctcc;s12:5’-tccctccctccc;s13:5’-tcccctcccctc;s14:5’-tccccctccccc;s15:5’-cccccctcccccc;
通过图2b可以说明在一定程度下,增加胞嘧啶的数目可以增加荧光强度,而且,胞嘧啶的位点也会影响到荧光强度,每三个胞嘧啶中间间隔一个脱氧核糖,荧光强度能达到最大强度。
图3a五条曲线分别是paa-agncs+ptk7+sgc8/s16、paa-agncs+sgc8/s16、paa-agncs+sgc8/s16、paa-agncs+ptk7和paa-agncs五种溶液的荧光强度曲线;b图是bsa,has,fn和ptk7四种蛋白质与sgc8/s16杂合双链dna作用之后的荧光强度。
图4a为不同ph条件下paa-agncs转化dna-agncs的荧光强度。b图为不同温度下paa-agncs转化dna-agncs的荧光强度。c图为不同反应时间paa-agncs转化dna-agncs的荧光强度。
图5为利用标准曲线法定量测定hela细胞中的ptk7,a图由下至上为一系列浓度梯度(0,0.03,0.1,0.2,0.4,0.6,0.8,1,1.25,1.5,1.7,and2.0nm)的ptk7荧光强度。b图为λem=650nm处荧光增加强度与ptk7浓度之间的线性关系。f和f0分别是hela细胞存在和不存在两种实验条件下的强度。
图6为利用标准曲线法测定定量测定ccrf-cem细胞中的ptk7,a图由下至上为一系列浓度梯度(0,0.05,0.1,0.2,0.3,0.4,0.6,0.8,1,1.5,1.7,and2.0nm)的ptk7荧光强度。b图为λem=650nm处荧光增加强度与ptk7浓度之间的线性关系。f和f0分别是ccrf-cem细胞存在和不存在两种实验条件下的强度。
具体实施方式
下面结合具体的实施例对本发明作进一步说明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,基于本发明的原理对本发明所做出的各种改动或修改同样落入本发明权利要求书所限定
实施例一:
步骤1.细胞的培育
选取海拉细胞(宫颈腺癌),ccrf-cem细胞(人类白血病细胞),拉莫斯细胞(人类伯基特淋巴瘤细胞)三种肿瘤细胞,利用含有10%的胎牛血清和100u/ml青霉素以及100μg/ml链霉素的dmem培养基在37℃的湿润气氛中含有5%的二氧化碳和95%的空气中进行培养。培养好之后,利用pbs清洗细胞,用于膜蛋白检测。
步骤2.paa保护的agncs的制备
室温避光搅拌条件下,取120μl1mmpaa溶液加入新配制的100μl10mmagno3中,加入适量的硼氢化钠还原溶液中的银离子,利用50mm磷酸盐缓冲剂调节溶液ph至7.4。混合溶液搅拌在黑暗中过夜,最终得到的溶液在双重蒸馏水中透析24h以除去未反应的银离子。得到的溶液在使用之前需要在避光条件下保存。
步骤3.agncs在paa与c-rich序列之间的转移反应
利用磷酸盐缓冲剂(10mm,ph7.4)配制2μl15nmdna溶液,加入到50μl已经制备好的paa保护的agncs溶液中,温度45℃下黑暗环境中搅拌15min。ph值通过在磷酸盐缓冲剂加入氢氧化钠或者硝酸调控到5.5。
实施例二:
分别将等体积等浓度50nm的sgc8和s16溶解到混合到ph7.4,含有10mmpbs和1mmmgcl2的磷酸盐缓冲剂中,溶液慢慢加热到90℃,然后大约1h冷却到室温,以确保核酸相互杂化完全,最终得到浓度为15nmdsdna。将目标膜蛋白ptk7溶解在磷酸盐缓冲剂,稀释到一定浓度梯度(0、0.1、0.2、0.4、0.6、0.8、1.0、1.25、1.5、1.7、2.0nm)的溶液,分别加入已配制好的dsdna溶液50μl,拆解杂化的核酸,确保能够完成纯化后的膜蛋白的定量检测,最终绘制ptk7-荧光强度标准曲线图。为了定量测定出膜蛋白在细胞膜上的表达,在室温下我们将dna杂合探针、细胞悬浮液(hela细胞或者ccrf-cem细胞)以及膜蛋白ptk7混合在一起(用量为2μl15nmdna溶液,ptk7加入量为10μl;细胞悬浮液约浓度105个/ml,取50μl。)37℃孵化1h,然后在1000rpm条件下离心5min除去细胞,在上清液中加入50μl已制备好的paa保护的agncs溶液,在45℃避光条件下孵化15min,稀释溶液至200μl,测试荧光吸收值在550nm处的荧光强度。
与现有的技术相比,现有技术太过于依赖复杂的机器,需要繁琐的预处理,如分离、净化等,大量的工作人工完成,利用该方法能够灵敏快速准确的进行原位膜蛋白的检测而且能够定量测量膜蛋白。这使得该方法在一些疾病的研究以及临床诊断上具有良好的应用前景。
sequencelisting
<110>中南大学
<120>一种膜蛋白检测制剂、非标记肿瘤纳米探针及使用方法
<130>无
<160>17
<170>patentinversion3.3
<210>1
<211>41
<212>dna
<213>核酸适配子为sgc8
<400>1
atctaactgctgcgccgccgggaaaatactgtacggttaga41
<210>2
<211>22
<212>dna
<213>富含胞嘧啶的单链s16
<400>2
tctaaccctccctcccgtacag22
<210>3
<211>12
<212>dna
<213>s1
<400>3
ttttcccctttt12
<210>4
<211>12
<212>dna
<213>s2
<400>4
tttccccccttt12
<210>5
<211>12
<212>dna
<213>s3
<400>5
ttcccccccctt12
<210>6
<211>12
<212>dna
<213>s4
<400>6
tcccccccccct12
<210>7
<211>12
<212>dna
<213>s5
<400>7
cccccccccccc12
<210>8
<211>12
<212>dna
<213>s6
<400>8
ttcctcctcctt12
<210>9
<211>12
<212>dna
<213>s7
<400>9
ttccttccttcc12
<210>10
<211>12
<212>dna
<213>s8
<400>10
cctttcctttcc12
<210>11
<211>14
<212>dna
<213>s9
<400>11
ccttttccttttcc14
<210>12
<211>16
<212>dna
<213>s10
<400>12
cctttttcctttttcc16
<210>13
<211>12
<212>dna
<213>s11
<400>13
tcctcctcctcc12
<210>14
<211>12
<212>dna
<213>s12
<400>14
tccctccctccc12
<210>15
<211>12
<212>dna
<213>s13
<400>15
tcccctcccctc12
<210>16
<211>12
<212>dna
<213>s14
<400>16
tccccctccccc12
<210>17
<211>13
<212>dna
<213>s15
<400>17
cccccctcccccc13
- 还没有人留言评论。精彩留言会获得点赞!