一种动物性食品中79种抗菌药物的筛查方法与流程
本发明属于生物检测
技术领域:
,具体涉及一种动物性食品中79种抗菌药物的筛查方法。
背景技术:
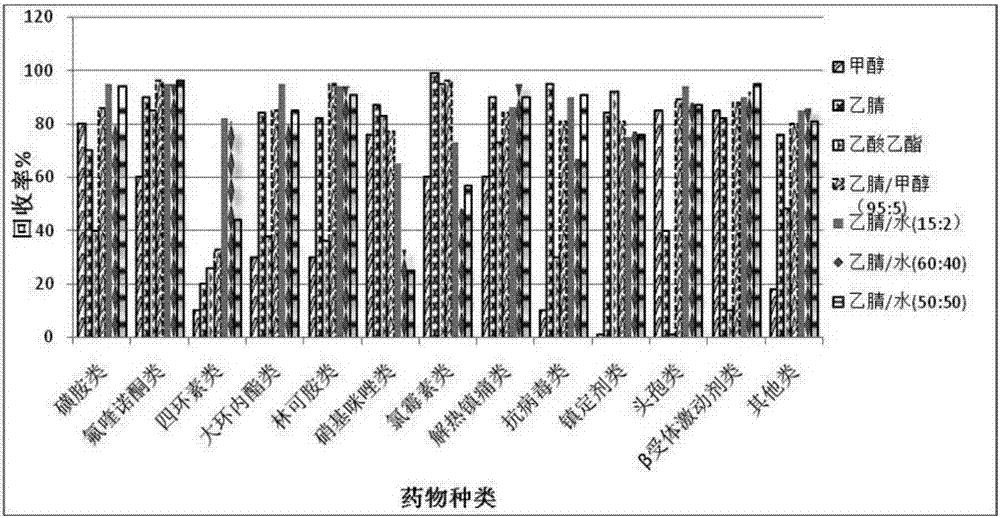
:近年来,我国的畜牧业一跃成为农业重要的支柱型产业,取得了巨大的成就,这与兽药的发展和合理使用是分不开的。兽药是预防、诊断、治疗动物疾病的重要物质,合理使用时,可降低畜禽的发病率与死亡率、促进动物生长、改善动物产品品质和提高饲料利用率。临床上常用的兽药有氟喹诺酮类、磺胺类、大环内酯类、四环素类、林可胺类等,但是目前在治疗和预防动物疾病过程中滥用药物情况比较普遍,一些药物用量已经远远超过了治疗和预防动物疾病的需要量。其毒性作用大多是通过长期积累而造成的,还可通过食物链进行传递,长期食用兽药残留超标的动物性食品可在人体产生过敏反应和细菌耐药性,具有潜在的致癌性,从而危害人类健康。中华人民共和国农业部公告第235号明确规定地西泮、地美硝唑、甲硝唑允许治疗使用,但是不得在动物性食品中检出;氯霉素、氨苯砜等禁止使用,不得在动物性食品中检出。并于2005年发布关于清查金刚烷胺等抗病毒药物的紧急通知,对兽用抗病毒药物的生产和使用进行严厉查处。兽药滥用现象在养殖环节是一种很普遍的现象,药物残留也大大影响了食品安全。β受体激动剂类药物在养殖环节应用更是一种不可忽视的问题,β-受体激动剂(β-agonists)是一类化学合成的苯乙醇胺类物质,在饲料中添加该类药物给动物饲喂后具有营养再分配作用,可以明显提高动物的瘦肉率,但是人们食用了这些药物残留的畜禽产品后会出现面色潮红、头痛、头晕、胸闷、心悸、四肢麻木等不良反应症状,严重的可能危及生命。因此国际上很多国家先后立法禁止在畜禽生产上使用该类药物,我国政府也明令禁止其使用。目前,欧盟和我国都将猪尿、猪肝、牛羊肉等动物性食品中该类药物的检测判定限定为1μg/l(μg/kg)。国内外现有的药物多残留检测方法主要有液相色谱法、液相色谱串联飞行时间质谱法和液相色谱串联质谱法等方法。现存的检测方法对单一种类药物研究已相对较为成熟,药物多残留的检测方法建立时需要考虑的因素较多,各药物的物理化学性质迥异,前处理方法较为复杂,存在灵敏度低、药物种类覆盖面窄等缺点。基于药物多残留技术具有的特殊监测和预警优势,近年来,我国各科研单位也逐渐重视药物多残留检测技术的研究。目前国内同时测定畜产品中多残留药物并对其准确定量的方法较为缺乏,因此亟需建立一种同时测定多种药物残留的检测技术。技术实现要素:本发明的目的在于:提供了一种动物性食品中79种抗菌药物的筛查方法,为畜产品及动物性食品添加残留抗菌药物提供了一种完整的检测方法,填充了国家行业残留检测标准,增添了抗菌药物筛查技术手段,为国家食品安全提供了安全保障。本发明的目的是以下述方式实现的:一种动物性食品中79种抗菌药物的筛查方法,具体包括以下步骤:所述79种抗菌药物为:(a)磺胺类:磺胺间甲氧嘧啶、磺胺甲恶唑、磺胺间二甲氧嘧啶、磺胺喹恶啉、磺胺二甲嘧啶、磺胺氯哒嗪、磺胺吡啶、磺胺二甲唑、磺胺甲噻二唑、磺胺二甲异嘧啶、磺胺氯吡嗪、磺胺甲氧嗪、磺胺邻二甲氧嘧啶、磺胺嘧啶、磺胺甲嘧啶、磺胺异噁唑、苯酰磺胺、磺胺对甲氧嘧啶、磺胺噻唑;(b)头孢类:头孢喹肟;(c)四环素类:土霉素、四环素、金霉素;(d)酰胺醇类:氯霉素、氟苯尼考、甲砜霉素;(e)大环内酯类:泰乐菌素、替米考星、阿奇霉素、克拉霉素、罗红霉素、吉他霉素a5;(f)林可胺类:林可霉素、克林霉素;(g)氟喹诺酮类:恩诺沙星、环丙沙星、沙拉沙星、二氟沙星、那氟沙星、诺氟沙星、培氟沙星、萘啶酸、西诺沙星、洛美沙星、氧氟沙星、恶喹酸、氟甲喹、依诺沙星;(h)硝基咪唑类:奥硝唑、替硝唑、地美硝唑、罗硝唑、甲硝唑、卡硝唑;(i)抗病毒类:金刚烷胺、金刚乙胺;(j)镇定剂类:氟哌利多、氟哌啶醇、阿扎哌隆、异丙嗪、地西泮;(k)解热镇痛类:对乙酰氨基酚、安替比林、氨基比林;(l)β-受体激动剂类:莱克多巴胺、克伦特罗、沙丁胺醇、溴布特罗、福莫特罗、拉贝特罗、喷布特罗、妥布特罗、西马特罗、齐帕特罗、氯丙那林;(m)其他类:阿托品、泰妙菌素、氨苯砜、甲氧苄啶;(1)标准工作溶液的配制:准确称取各标准品10mg,于10ml量瓶中,用甲醇溶解并稀释至刻度,配制成1mg/ml各标准贮备液;对于难溶药物,恩诺沙星、环丙沙星、诺氟沙星加入少量0.1mol/lhcl溶液,洛美沙星加入少量0.1mol/lnaoh溶液,头孢喹肟加适量二甲基亚砜使其溶解后,用甲醇定至刻度;量取各标准贮备液至100ml量瓶中,用甲醇稀释至刻度,制得混合标准中间溶液;其中磺胺类、氟喹诺酮类、林可胺类、氨苯砜、泰妙菌素和甲氧苄啶各取0.4ml;解热镇痛类和金刚乙胺各取2ml;头孢喹肟取4ml;四环素类、泰乐菌素、替米考星、吉他霉素a5、奥硝唑和金刚乙胺各取1ml;β-受体激动剂类、酰胺醇类、阿托品、氟哌利多和氟哌啶醇各取0.2ml;阿扎派隆、异丙嗪和地西泮各取0.1ml;移取混合标准中间溶液0.1ml、0.2ml、0.5ml分别至10ml量瓶中,用甲醇稀释至刻度,制得loq浓度混合标准工作溶液、2loq浓度混合标准工作溶液和5loq浓度混合标准工作溶液;(2)待测样品的分析前处理:1)提取:称取均质试样2±0.02g于50ml聚丙烯离心管中,先后加入na2edta溶液和乙腈、甲醇、乙酸乙酯溶液,涡旋10-30s混匀,振荡10-30min;以5000-8000r/min转速离心5min,上清液转移至50ml聚丙烯离心管中;加入提取液对残渣进行重复提取,合并提取液;并于40-50℃下氮吹至溶液剩余约1-2ml;作为备用液;2)净化:采用agelacleanertpep-2固相萃取柱分别用5-8ml甲醇和3-6ml水活化,将备用液全部过柱,控制流速为约1-3滴/s;用淋洗液淋洗,真空抽干3-5min;用洗脱液洗脱,洗脱液于30-40℃下氮气吹干;准确加入20%乙腈水溶液1-3ml溶解定容,涡旋混匀后过0.22μm微孔尼龙滤膜;(3)标准曲线的绘制:精密量取1μg/ml混合标准溶液适量,空白试样基质溶液配制成浓度为1、2、5、10、20、50、100ng/ml的系列标准溶液,供液相色谱-串联质谱测定。以特征离子质量色谱峰面积为纵坐标,相对应的标准溶液浓度为横坐标,绘制标准曲线,获得79种抗菌药物的定性分析色谱图库;(4)对步骤(3)所述的含有各个药物定量限loq的2loq浓度的试样进行质谱分析,获得各个药物的准分子离子,以及两个特征离子:定性离子和定量离子,如下表所示:(5)定性分析:对经过步骤(2)所述方法处理的待测样品选择1个母离子和2个特征离子,进行一次性液相色谱分析,样品中待测物的保留时间与步骤(3)所得谱图库中对应的保留时间偏差在±2.5%之内,且样品中各组分定性离子的相对丰度与浓度接近的标准溶液中对应的定性离子的相对丰度进行比较,偏差不超过下表规定的范围,则可判定为样品中存在对应的待测物:相对离子丰度(%)>50>20~50>10~20≤10允许的最大偏差(%)±20±25±30±50(6)定量分析:在仪器最佳工作条件下,取经过步骤(2)所述方法处理的待测样品和步骤(1)所制备的混合标准工作液进行质谱分析,得到色谱峰面积响应值,作单点或多点校准,用外标法定量;待测样品溶液中待测物的响应值和混合标准工作也中各待测物的特征离子的响应值均应在仪器测定的线性范围内,若待测样品溶液浓度超过线性范围时,用乙腈/水(v/v,20:80)溶液稀释后重新测定;试样中各药物含量x以质量分数计,单位为毫克每千克(mg/kg),按公式(1)计算:式中:a—待测样品溶液中待测物的峰面积;as—混合标准工作液中相应待测物的峰面积;cs—混合标准工作液中待测物浓度,单位为微克每毫升(μg/ml);v—溶解残余物的体积,单位为毫升(ml);m—样品的质量,单位为克(g);v1—过固相萃取柱的提取液体积,单位为毫升(ml);v2—总提取液体积,单位为毫升(ml);n—稀释倍数。测定结果用平行测定的算术平均值表示,结果保留三位有效数字。所述动物性食品为动物肌肉类食品、动物肝脏、动物肾脏、鸡蛋和奶类制品。所述步骤(3)和步骤(5)中的液相色谱分析条件为:色谱柱:watersuplcc18,100mm×2.1mmid,1.7μm;流动相a:水溶液,流动相b:乙腈溶液,流动相c:0.1%甲酸水溶液,流动相d:甲醇溶液;下表为正、负离子梯度洗脱方式;流速:0.1-0.3ml/min;进样量:2μl;柱温:25-40℃;注:6为线性变化,1为即时变化。所述步骤(4)和步骤(6)中的质谱分析条件为:所述步骤(2)中的振荡时间为20min,提取液为乙腈和水的混合溶液,体积比为15:2;淋洗液为水1-3ml,洗脱液为乙腈溶液和甲醇溶液各2-5ml。所述步骤(2)中na2edta溶液的浓度为0.1mol/l。本方法针对我国畜产品养殖过程中常用的抗菌类、抗病毒类、解热镇痛类、镇定剂类、β-受体激动剂类等药物,基于超高效液相色谱串联质谱具有的高效分离性和可同时准确定性定量的特点,建立了同时测定牛肉、鸡肉中磺胺类、氟喹诺酮类、四环素类、硝基咪唑类、头孢类、酰胺醇类、大环内酯类、β-受体激动剂类和林可胺类等13类79种药物的uplc-ms/ms方法,可为主管部门监管市场上含有超标兽药及违禁药物的畜产品提供了技术保障,对畜产品饲养中非法、违规使用药物的行为给予重磅一击。相对于现有技术,本发明搭建的高通量筛查和定量的检测平台,使得在本领域内从检测单一化合物向同时检测多种化合物的方向发展,与原有的技术相比,检测成本仅为原有的二分之一左右,样品的检测周期缩短为原来的一半,极大提高了工作效率。本发明建立筛查技术在农业部实施的兽药残留监控计划、无公害畜产品检测计划以及全国畜产品例行检测计划等活动中发挥了有效的作用。在我国省级畜产品质检中心进行了样品检测,据不完全统计,推广应用样品数量共计2万余份。从实验成本来说,使用高通量筛查方法,大大降低了检测成本,缩短了一半的检测时间,经济效益每年可节省约800万元。本发明的应用为我国动物及动物产品的质量安全提供强有力的技术保障;为我国的动物及动物产品进入国际市场,冲破“绿色贸易壁垒”的限制提供保障;提高我国在国际贸易中的竞争地位,保证我国农产品的安全出口,增加外汇收入,创造良好的经济效益;同时对保证食品生产行业的健康发展,促进经济的持续发展,维护社会稳定,产生巨大社会效益。附图说明图1是不同提取液的对各药物回收率的影响。图2是不同固相萃取柱对各药物回收率的影响。图3是不同洗脱液对各药物回收率的影响。图4是磺胺吡啶的色谱图。图5是磺胺嘧啶的色谱图。图6是磺胺甲恶唑的色谱图。图7是磺胺噻唑的色谱图。图8是磺胺甲嘧啶的色谱图。图9是磺胺二甲唑和磺胺异噁唑的色谱图,其中左侧峰为磺胺二甲唑,右侧峰为磺胺异噁唑。图10是磺胺甲噻二唑的色谱图。图11是苯酰磺胺的色谱图。图12是磺胺二甲异嘧啶的色谱图。图13是磺胺二甲嘧啶的色谱图。图14是磺胺对甲氧嘧啶、磺胺甲氧嗪和磺胺间甲氧嘧啶的色谱图,其中左侧峰为磺胺对甲氧嘧啶,中间峰为磺胺甲氧嗪,右侧峰为磺胺间甲氧嘧啶。图15是磺胺氯哒嗪和磺胺氯吡嗪的色谱图,其中左侧峰为磺胺氯哒嗪,右侧峰为磺胺氯吡嗪。图16是甲氧苄啶的色谱图。图17是磺胺邻二甲氧嘧啶和磺胺间二甲氧嘧啶的色谱图,其中左侧峰为磺胺邻二甲氧嘧啶,右侧峰为磺胺间二甲氧嘧啶。图18是磺胺喹恶啉的色谱图。图19是地西泮的色谱图。图20是地美硝唑的色谱图。图21是头孢喹肟的色谱图。图22是甲硝唑的色谱图。图23是罗硝唑的色谱图。图24是奥硝唑的色谱图。图25是替硝唑的色谱图。图26是金刚烷胺的色谱图。图27是金刚乙胺的色谱图。图28是对乙酰氨基酚的色谱图。图29是氨基比林的色谱图。图30是安替比林的色谱图。图31是氨苯砜的色谱图。图32是阿托品的色谱图。图33是泰妙菌素的色谱图。图34是萘啶酸的色谱图。图35是恶喹酸的色谱图。图36是氟甲喹的色谱图。图37是西诺沙星的色谱图。图38是诺氟沙星的色谱图。图39是伊诺沙星的色谱图。图40是环丙沙星的色谱图。图41是培氟沙星的色谱图。图42是洛美沙星的色谱图。图43是恩诺沙星的色谱图。图44是那氟沙星的色谱图。图45是氧氟沙星的色谱图。图46是沙拉沙星的色谱图。图47是二氟沙星的色谱图。图48是异丙嗪的色谱图。图49是林可霉素的色谱图。图50是克林霉素的色谱图。图51是土霉素的色谱图。图52是金霉素的色谱图。图53是四环素的色谱图。图54是克拉霉素的色谱图。图55是阿奇霉素的色谱图。图56是吉他霉素的色谱图。图57是罗红霉素的色谱图。图58是替米考星的色谱图。图59是泰乐菌素的色呕吐。图60是氟哌啶醇的色谱图。图61是氟哌利多的色谱图。图62是阿扎哌隆的色谱图。图63是卡硝唑的色谱图。图64是氯霉素的色谱图。图65是甲砜霉素的色谱图。图66是氟苯尼考的色谱图。图67是克伦特罗的色谱图。图68是沙丁胺醇的色谱图。图69是喷布特罗的色谱图。图70是妥布特罗的色谱图。图71是拉贝特罗的色谱图。图72是福莫特罗的色谱图。图73是西马特罗的色谱图。图74是氯丙那林的色谱图。图75是溴布特罗的色谱图。图76是莱克多巴胺的色谱图。图77是齐帕特罗的色谱图。具体实施方式所检药物共13类79种抗菌药物,分别为:(a)磺胺类:磺胺间甲氧嘧啶、磺胺甲恶唑、磺胺间二甲氧嘧啶、磺胺喹恶啉、磺胺二甲嘧啶、磺胺氯哒嗪、磺胺吡啶、磺胺二甲唑、磺胺甲噻二唑、磺胺二甲异嘧啶、磺胺氯吡嗪、磺胺甲氧嗪、磺胺邻二甲氧嘧啶、磺胺嘧啶、磺胺甲嘧啶、磺胺异噁唑、苯酰磺胺、磺胺对甲氧嘧啶、磺胺噻唑;(b)头孢类:头孢喹肟;(c)四环素类:土霉素、四环素、金霉素;(d)酰胺醇类:氯霉素、氟苯尼考、甲砜霉素;(e)大环内酯类:泰乐菌素、替米考星、阿奇霉素、克拉霉素、罗红霉素、吉他霉素a5;(f)林可胺类:林可霉素、克林霉素;(g)氟喹诺酮类:恩诺沙星、环丙沙星、沙拉沙星、二氟沙星、那氟沙星、诺氟沙星、培氟沙星、萘啶酸、西诺沙星、洛美沙星、氧氟沙星、恶喹酸、氟甲喹、依诺沙星;(h)硝基咪唑类:奥硝唑、替硝唑、地美硝唑、罗硝唑、甲硝唑、卡硝唑;(i)抗病毒类:金刚烷胺、金刚乙胺;(j)镇定剂类:氟哌利多、氟哌啶醇、阿扎哌隆、异丙嗪、地西泮;(k)解热镇痛类:对乙酰氨基酚、安替比林、氨基比林;(l)β-受体激动剂类:莱克多巴胺、克伦特罗、沙丁胺醇、溴布特罗、福莫特罗、拉贝特罗、喷布特罗、妥布特罗、西马特罗、齐帕特罗、氯丙那林;(m)其他类:阿托品、泰妙菌素、氨苯砜、甲氧苄啶。1.标准工作溶液的配制:准确称取各标准品10mg,于10ml量瓶中,用甲醇溶解并稀释至刻度,配制成1mg/ml各标准贮备液。-20℃下保存,有效期为3个月;对于难溶药物,恩诺沙星、环丙沙星、诺氟沙星加入少量0.1mol/lhcl溶液,洛美沙星加入少量0.1mol/lnaoh溶液,头孢喹肟加适量二甲基亚砜使其溶解后,用甲醇定至刻度;量取各标准贮备液至100ml量瓶中,用甲醇稀释至刻度,制得混合标准中间溶液;其中磺胺类、氟喹诺酮类、林可胺类、氨苯砜、泰妙菌素和甲氧苄啶各取0.4ml;解热镇痛类和金刚乙胺各取2ml;头孢喹肟取4ml;四环素类、泰乐菌素、替米考星、吉他霉素a5、奥硝唑和金刚乙胺各取1ml;β-受体激动剂类、酰胺醇类、阿托品、氟哌利多和氟哌啶醇各取0.2ml;阿扎派隆、异丙嗪和地西泮各取0.1ml;移取混合标准中间溶液0.1ml、0.2ml、0.5ml分别至10ml量瓶中,用甲醇稀释至刻度,制得loq浓度混合标准工作溶液、2loq浓度混合标准工作溶液和5loq浓度混合标准工作溶液,4℃避光保存,有效期1周。2.待测样品的分析前处理:2.1制备:牛肉、鸡肉预先切碎后于组织匀浆机中匀浆,作为备用样品,将备用样品于-20℃下保存。2.2提取:肌肉:称取均质试样2±0.02g于50ml聚丙烯离心管中,先后加入200μl0.1mol/lna2edta溶液和10ml乙腈/甲醇(v/v,95:5)提取液,涡旋10-30s混匀,振荡10-30min;以5000-8000r/min转速离心5min,上清液转移至50ml聚丙烯离心管中;加入10ml乙腈/水(v/v,15:2)溶液进行重复提取,合并提取液;并于40-50℃下氮吹至溶液剩余约0.5-1ml;分别用0.5ml乙腈和7.5ml水清洗,作为备用液。肝脏、肾脏、鸡蛋、牛奶:称取2g试样(±0.02g)于50ml离心管中,分别加入200μl0.1mol/ledta溶液和10ml乙腈,涡旋混匀后,振荡20min。于离心机上5000r/min离心5min,将上清液转移至50ml离心管中。向残渣中加入10ml乙腈/水(v/v,15:2)溶液进行重复提取,合并两次提取液;并于40-50℃下氮吹至溶液剩余约0.5-1ml;分别用0.5ml乙腈和7.5ml水清洗,作为备用液。本发明分别考察了乙腈、甲醇、乙酸乙酯溶液的提取效果,而后又分别对乙腈/甲醇(95:5,v/v)、乙腈/水(15:2,v/v)、乙腈/水(60:40,v/v)和乙腈/水(50:50,v/v)溶液等提取液进行考察,图1为不同提取液的对各药物回收率的影响。实验结果表明:甲醇和乙酸乙酯对大部分药物均有较好的提取效率,但是对大环内酯类、林可胺类、抗病毒类提取效率较差;乙腈的提取效果较好,乙腈中加入少量甲醇后使提取效率更佳;以上四种纯有机体系对四环素类药物提取率在10%~26%之间。乙腈/水(15:2,v/v)对各个药物均有较好的提取效率,特别是对四环素类药物的提取效果相对较高,但是对氯霉素和部分镇定剂类药物的提取效率比乙腈/甲醇(95:5,v/v)稍低;乙腈/水(60:40,v/v)和乙腈/水(50:50,v/v)溶液可提取出较多的极性杂质,四环素类、大环内酯类、解热镇痛类药物均有较高的基质抑制,抑制率达80%。乙腈/甲醇(95:5,v/v)和乙腈/水(15:2,v/v)具有较好的互补作用,分别使用这两种溶液时各药物具有相对较高的回收率,且具有较低的基质效应,杂峰干扰少。含水量过多不利于提取液的浓缩,但提取液中加少量na2edta溶液和少量水能明显提高大环内酯类和四环素类药物的回收率,可能是因为这两类药物在溶液中易形成二价或三价阳离子从而在提取阶段造成损失,na2edta可以与这两类药物形成络合物,因而可避免因流失而造成的回收率偏低。本发明还对提取过程中的震荡时间做了考察,比较了震荡10min和20min各药物的提取结果。结果表明震荡时间延长后,除地美硝唑和对乙酰氨基酚回收率有10%降低外,其他药物的回收率有0~25%的提高,故在提取阶段选择20min作为震荡时间。2.3净化:采用oasishlb、agelacleanertpep-2、varianbondelutc18和waterssep-pakc18固相萃取柱分别用5ml甲醇和5ml水活化,将备用液全部过柱,控制流速为约1-3滴/s;用2ml淋洗液淋洗,真空抽干3-5min;用3ml乙腈和3ml甲醇分别洗脱,收集洗脱液于40℃下氮气吹干;准确加入20%乙腈水溶液0.5ml溶解定容,涡旋混匀后过0.22μm微孔尼龙滤膜;经过对固相萃取柱的筛选,通过图2比较发现c18柱对磺胺类、大环内酯类药物的回收效果稍差,且柱床干涸易造成柱床塌陷,不能保证结果的稳定性;hlb柱和pep-2柱对各药物均有较好的保留和净化能力,具有相似的回收效果。hlb固相萃取柱价格较贵,pep-2柱价格相对较低,在具有相同净化效果的条件下,故选择agelacleanertpep-2固相萃取柱为本发明的净化柱。洗脱液的种类直接影响固相萃取柱的净化效果,本发明对洗脱液进行了考察,分别使用乙腈、甲醇、乙腈/甲酸水(ph3.5)(2:1,v/v)和乙腈/甲醇(1:1,v/v)为洗脱液,图3选择了磺胺噻唑、磺胺甲噻二唑等对洗脱液种类较为敏感的药物做代表,显示各洗脱液的洗脱能力。结果表明,总体来说乙腈的洗脱效果不如甲醇,但是甲醇对甲硝唑、地美硝唑和林可霉素的洗脱效果不如乙腈;乙腈/甲酸水(ph3.5)(2:1,v/v)为洗脱液时,苯酰磺胺的回收率仅为30%;当使用3ml乙腈和3ml甲醇作为洗脱液分别洗脱时,各个药物的回收率最优,且具有较好峰形。本发明对淋洗液也做了考察,头孢类药物对乙腈、甲醇等有机溶剂较为敏感,低浓度的乙腈水溶液和甲醇水溶液即可将其洗出,故使用2ml水为淋洗液。3.液相色谱条件:色谱柱:watersuplcc18(100mm×2.1mmid,1.7μm);流动相a:水溶液,流动相b:乙腈溶液,流动相c:0.1%甲酸水溶液,流动相d:甲醇溶液;表1为正、负离子梯度洗脱方式;流速:0.1-0.3ml/min;进样量:2μl;柱温:25-40℃。表1梯度洗脱程序注:6为线性变化,1为即时变化。经过液相条件的反复优化,多种组分的同时分析时,为了增大各药物间的分离度,常采用梯度洗脱方式。本方法以watersc18(100mm×2.1mmid.,1.7μm)为色谱柱,考察了甲醇、乙腈、水和0.1%甲酸水溶液所组成的四种流动相体系,经过对比各药物的色谱峰响应及峰型,发现正离子模式下使用甲醇时各药物的s/n较高,而0.1%甲酸水能使大部分药物的离子化效率有所提高,部分硝基咪唑类药物在甲醇-水为流动相时具有较高的效应和较好的峰型[12],为了兼顾所有药物的测定,正离子模式下使用的流动相为甲醇-0.1%甲酸水溶液;负离子模式下使用乙腈-水作为流动相时酰胺醇类药物的色谱峰型和响应值均为最优。4.质谱条件离子源参数、质谱方法参数见表2:表2离子源参数经过质谱条件的筛选与优化,方法使用20%乙腈水溶液对各标准溶液进行稀释,制得200ng/ml各个药物的标准溶液,在esi正、负离子模式下,采用流动注射泵连续进样方式进行全扫描,确定各药物的母离子。并对该母离子下的子离子全扫描,在mrm模式下对其质谱条件进行优化。最后选取丰度较强的两个子离子作为定性、定量离子。其中磺胺类、氟喹诺酮类、林可胺类、四环素类、硝基咪唑类、大环内酯类等药物在[m+h]+模式下灵敏度较高,故选择在正离子模式下进行测定;酰胺醇类药物在[m-h]-模式下具有较高响应值,故选择在负离子模式下进行测定。5.色谱图库的建立:精密量取1μg/ml混合标准溶液适量,空白试样基质溶液配制成浓度为1、2、5、10、20、50、100ng/ml的系列标准溶液,供液相色谱-串联质谱测定。以特征离子质量色谱峰面积为纵坐标,相对应的标准溶液浓度为横坐标,绘制标准曲线,获得79种抗菌药物的定性分析色谱图库,如图4-图77所示;对步骤5所述的含有各个药物定量限loq的2loq浓度的试样进行质谱分析,离子源参数、质谱方法参数见表2,获得各个药物的准分子离子,以及两个特征离子:定性离子和定量离子及相应的碰撞能量参考值,如表3所示:表3药物的定性、定量离子和锥孔电压、碰撞能定性分析:对经过步骤2所述方法处理的待测样品选择1个母离子和2个特征离子,进行一次性液相色谱分析,样品中待测物的保留时间与步骤5所得谱图库中对应的保留时间偏差在±2.5%之内,且样品中各组分定性离子的相对丰度与浓度接近的标准溶液中对应的定性离子的相对丰度进行比较,偏差不超过表4规定的范围,则可判定为样品中存在对应的待测物:表4定性确证时相对离子丰度的最大允许偏差相对离子丰度(%)>50>20~50>10~20≤10允许的最大偏差(%)±20±25±30±50定量分析:取经过步骤(2)所述方法处理的待测样品和步骤(1)所制备的混合标准工作液进行质谱分析,得到色谱峰面积响应值,作单点或多点校准,用外标法定量;待测样品溶液中待测物的响应值均应在仪器测定的线性范围内,若待测样品溶液浓度超过线性范围时,用乙腈/水(v/v,20:80)溶液稀释后重新测定;试样中各药物含量x以质量分数计,单位为毫克每千克(mg/kg),按公式(1)计算:式中:a—待测样品溶液中待测物的峰面积;as—混合标准工作液中相应待测物的峰面积;cs—混合标准工作液中待测物浓度,单位为微克每毫升(μg/ml);v—溶解残余物的体积,单位为毫升(ml);m—样品的质量,单位为克(g);v1—过固相萃取柱的提取液体积,单位为毫升(ml);v2—总提取液体积,单位为毫升(ml);n—稀释倍数。测定结果用平行测定的算术平均值表示,结果保留三位有效数字。方法学验证8.1.线性:称取空白试样,按照步骤2所述方法处理,洗脱液在氮气吹干前,准确移取混合标准溶液适量,加入洗脱液中,制得各个药物定量限loq的0.5loq、loq、2loq、5loq、10loq、20loq浓度的基质匹配标准溶液,各药物在相关浓度范围内呈良好线性相关,如表5-6,相关系数均大于0.99。8.2.检测限和定量限检测限:添加一定量的混合标准溶液于2g空白样品中,经过提取净化并测定,药物的信噪比s/n≥3时,即可得到药物的检测限,其结果见表5-6。定量限:添加一定量的混合标准溶液于2g空白样品中,经过提取净化并测定,药物的信噪比s/n≥10时,即可得到药物的定量限,其结果见表5-6。8.3.方法精密度与准确度在牛、鸡肌肉空白组织中分别添加loq、2loq、5loq三个浓度混合标准溶液,每个浓度做5个平行。结果表明,牛肉中79种药物的平均回收率、变异系数如表5所示。可以看出,79种药物的平均回收率在45%-125%之间,批内相对标准偏差均小于20%。鸡肉中79种药物的平均回收率、变异系数如表6所示。可以看出,79种药物的平均回收率在43%-118%之间,批内相对标准偏差均小于20%。表5牛肉中79种药物的线性范围、相关系数、平均回收率及rsd表6鸡肉中79种药物的线性范围、相关系数、平均回收率及rsd9.小结9.1方法灵敏度本发明在猪、牛、羊、鸡的肌肉组织、鸡蛋、牛奶中阿扎派隆、异丙嗪和地西泮的定量限为0.5μg/kg;β-受体激动剂类、酰胺醇类、阿托品、氟哌利多和氟哌啶醇的定量限为1μg/kg;磺胺类、氟喹诺酮类、林可胺类、氨苯砜、泰妙菌素和甲氧苄啶的定量限为2μg/kg;四环素类、泰乐菌素、替米考星、吉他霉素a5、奥硝唑和金刚乙胺的定量限为5μg/kg;解热镇痛类和金刚烷胺的定量限为10μg/kg;头孢喹肟的定量限为20μg/kg。在猪、牛、羊、鸡的肝脏组织、肾脏组织中阿扎派隆、异丙嗪和地西泮的定量限为1μg/kg;β-受体激动剂类、酰胺醇类、阿托品、氟哌利多和氟哌啶醇的定量限为2μg/kg;磺胺类、氟喹诺酮类、林可胺类、氨苯砜、泰妙菌素和甲氧苄啶的定量限为4μg/kg;四环素类、泰乐菌素、替米考星、吉他霉素a5、奥硝唑和金刚乙胺的定量限为10μg/kg;解热镇痛类和金刚烷胺的定量限为20μg/kg;头孢喹肟的定量限为40μg/kg。9.2准确度本发明在猪、牛、羊、鸡的肌肉组织、鸡蛋、牛奶中0.5~20μg/kg添加浓度水平上的回收率为30%~120%,在肝脏组织、肾脏组织中1~40μg/kg添加浓度水平上的回收率为30%~120%。9.3精密度本发明的批内相对标准偏差≤20%,批间相对标准偏差≤20%。10.实际样品检测如表7所示:表7实际样品检测结果以上检测数据采用uplc-ms/ms技术建立了猪、牛、羊、鸡的肌肉组织、鸡蛋、牛奶中79种药物残留的快速筛查分析方法。该方法与传统畜产品中单一药物种类检测方法相比,检测效率可提高5倍以上,每天可处理45批样品,并能对检测结果进行准确定性定量,相应检测成本可降低60%左右,并具有快速、准确、灵敏度高、筛查范围广等特点,适合于畜产品中兽药残留的快速筛查分析,为畜产品进行风险监测提供了一种有力的技术。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1