一种芪胶升白胶囊的检测方法与流程
本发明涉及一种芪胶升白胶囊的检测方法,属于药品技术的领域;
背景技术
芪胶升白胶囊是一种苗医中医互补的民族药,主要由大枣、阿胶、血人参、淫羊藿、苦参、黄芪、当归等组成,具有补血益气的功效,临床上用于治疗气血亏损所引起的头昏眼花、气短乏力、自汗盗汗,以及白细胞减少症等。
目前,芪胶升白胶囊执行的标准为国家食品药品监督管理局标准(编号ws-10026(zd-0026-2002-2012z)),在该标准中,仅对当归、黄芪、苦参、淫羊藿进行定性鉴别,对淫羊藿苷进行定量检查。但是芪胶升白胶囊作为7味中药材组成的中药复方制剂,组方成分复杂,按照目前的质量标准,是不能更好的控制其质量标准,提升产品的内在质量的。
技术实现要素:
本发明的目的在于,提供一种芪胶升白胶囊的检测方法,所述的检测方法准确,专属性强,可以作为芪胶升白胶囊胶囊的质量控制方法。本发明对芪胶升白胶囊中对大枣、当归、苦参、黄芪和淫羊藿的进行薄层鉴别;对苦参碱和/或淫羊藿苷进行含量测定,增加大枣薄层色谱鉴别;测定苦参药材中苦参碱的量,为建立有效可靠的质量标准,能够准确反映中药整体质量提供数据参考。能更好地控制该产品的治疗,确保其临床疗效。
为解决上述技术问题,本发明采用如下技术方案实现:一种芪胶升白胶囊的检测方法,所述药物由大枣240-260份、阿胶240-250份、血人参365-385份、淫羊藿365-385份、苦参240-260份、黄芪740-760份和当归240-260份按照下述方法制备而成:以上七味,除阿胶外,当归粉碎成细粉;其余大枣等五味,加水煎煮三次,第一次2小时,第二次1.5小时,第三次1小时,合并煎液,滤过,滤液加入烊化后的阿胶,搅匀,浓缩至75-85℃下相对密度为1.30的稠膏,加入上述当归细粉,混匀,制成颗粒,干燥,装入胶囊,即得;所述检测方法包括鉴别方法和含量测定方法;所述鉴别方法包括对大枣的薄层鉴别、当归的薄层鉴别、苦参的薄层鉴别、黄芪的薄层鉴别和/或淫羊藿的薄层鉴别;所述含量测定方法包括对苦参碱和/或淫羊藿苷的含量测定。
前述芪胶升白胶囊的检测方法中,所述大枣的薄层鉴别为:取药物颗粒内容物3g,加乙酸乙酯30ml,超声处理30分钟,过滤,蒸干滤液,残渣加甲醇0.5ml使溶解,作为供试品溶液;另取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,膏加乙酸乙酯30ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,作为对照药材溶液,吸取供试品溶液5μl和对照药材溶液10μl,分别点于同一硅胶h薄层板上,以甲苯-乙酸乙酯-冰醋酸=60:6:0.05为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。
前述芪胶升白胶囊的检测方法中,所述当归的薄层鉴别方法为:取药物颗粒内容物5g,加正己烷20ml,超声处理20分钟,滤过,滤液挥至1ml,作为供试品溶液;另取当归对照药材0.5g,同法制成对照药材溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取上述两种溶液各5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯=9:1为展开剂,展开,取出,晾干,置波长为365nm紫外灯下检视,供试品色谱中,在于对照药材色谱相应的位置上,先相同颜色的荧光斑点;
所述苦参的薄层鉴别方法为:取药物颗粒内容物1g,加乙醇30ml,加热回流30分钟,放冷,滤过,滤液蒸干残渣加水20ml使溶解,滤过,滤液置分液漏斗中,加浓度为25%-28%的浓氨试液0.5ml,用三氯甲烷振摇提取2次,每次10ml,合并三氯甲烷液,蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液;另取苦参碱和槐定碱对照品,分别加无水乙醇制成每1ml含1mg的溶液,作为对照品溶液,照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取上述三种溶液各5μl,分别点于同一硅胶g薄层板上,以环己烷-丙酮-乙酸乙酯-浓氨试液=2:3:4:0.5为展开剂,置氨蒸气预饱和的展缸内,预饱和30分钟,展开,取出,晾干,喷以稀碘化铋钾试液;供试品色谱中,在于对照品色谱相应的位置上,显相同颜色的斑点;
所述黄芪的薄层鉴别方法为:取药物颗粒内容物5g,加60-90℃的石油醚30ml,超声处理20分钟,滤过,药渣挥尽石油醚,加甲醇50ml,加热回流30分钟,滤过,滤渣用少量甲醇洗涤,洗液与滤液合并,蒸干,残渣加水10ml,加热使溶解,放冷,置离心管中离心10分钟,取上清液,用三氯甲烷振摇提取2次,每次15ml,弃去三氯甲烷液,用以水饱和的正丁醇振摇提取3次,第一次加10ml,第二次加10ml,第三次加5ml,合并正丁醇液,加氨试液洗涤3次,每次15ml,弃去氨液;正丁醇液用以正丁醇饱和的水洗至中性,弃去水液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取供试品溶液10μl、对照品溶液5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水=13:7:2,10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;再置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑;
淫羊藿的薄层鉴别方法为:取药物颗粒内容物5g,加60-90℃的石油醚30ml,超声处理20分钟,滤过,药渣挥尽石油醚,加甲醇50ml,加热回流30分钟,滤过,滤渣用5ml甲醇洗涤,洗液与滤液合并,蒸干,残渣加水10ml,加热使溶解,放冷,置离心管中离心10分钟,取上清液,用三氯甲烷振摇提取2次,每次15ml,弃去三氯甲烷液,用以水饱和的正丁醇振摇提取3次,第一次加10ml,第二次加10ml,第三次加5ml,合并正丁醇液,加氨试液洗涤3次,每次15ml,弃去氨液;正丁醇液用以正丁醇饱和的水洗至中性,弃去水液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;取淫羊藿苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取供试品溶液2~4μl和对照品溶液5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水=13:7:2,10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;再置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。
前述芪胶升白胶囊的检测方法中,所述苦参碱的含量测定方法为:照高效液相色谱法(中国药典2015年版四部0512法)测定:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:甲醇为a相,0.1%三乙胺为b相,洗脱条件:53%a-47%b;检测波长为220nm;理论塔板数按苦参碱峰计算应不低于4000;
对照品溶液的制备:精密称取苦参碱对照品,加甲醇制成每1ml含0.2mg的溶液,摇匀,即得;
供试品溶液的制备:取药物颗粒内容物1.0g,精密称定,置具塞锥形瓶中,加入浓度为25%-28%的浓氨试液2ml润湿,加入三氯甲烷30ml,加热回流1.5小时,过滤,滤液蒸干,残渣加10ml甲醇溶解,摇匀,即得;
测定法:分别吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。
前述芪胶升白胶囊的检测方法中,所述淫羊藿苷的含量测定方法为:照高效液相色谱法(中国药典2010年版一部附录ⅵd)测定:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:乙腈为a相,水为b相,洗脱条件:25.5%a-74.5%b;检测波长为270nm;理论塔板数按淫羊藿苷峰计算应不低于3000;
对照品溶液的制备:取淫羊藿苷对照品,精密称定,加甲醇制成每1ml含25μg的溶液,即得;
供试品溶液的制备:取药物内容物,研细后,取1.5g,精密称定,置具塞锥形瓶中,精密加入70%乙醇25ml,密塞,称定重量,超声处理,1小时,超声功率250w,超声频率35khz,取出,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
前述芪胶升白胶囊的检测方法中,所述药物这样检测:
(1)性状:硬胶囊内容物为棕褐色的颗粒及粉末;味苦;
(2)鉴别:包括对大枣的薄层鉴别、当归的薄层鉴别、苦参的薄层鉴别、黄芪的薄层鉴别和/或淫羊藿的薄层鉴别;
所述大枣的薄层鉴别为:取药物颗粒内容物3g,加乙酸乙酯30ml,超声处理30分钟,过滤,蒸干滤液,残渣加甲醇0.5ml使溶解,作为供试品溶液;另取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,膏加乙酸乙酯30ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,作为对照药材溶液,吸取供试品溶液5μl和对照药材溶液10μl,分别点于同一硅胶h薄层板上,以甲苯-乙酸乙酯-冰醋酸=60:6:0.05为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点;
所述当归的薄层鉴别方法为:取药物颗粒内容物5g,加正己烷20ml,超声处理20分钟,滤过,滤液挥至1ml,作为供试品溶液;另取当归对照药材0.5g,同法制成对照药材溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取上述两种溶液各5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯=9:1为展开剂,展开,取出,晾干,置波长为365nm紫外灯下检视,供试品色谱中,在于对照药材色谱相应的位置上,先相同颜色的荧光斑点;
所述苦参的薄层鉴别方法为:取药物颗粒内容物1g,加乙醇30ml,加热回流30分钟,放冷,滤过,滤液蒸干残渣加水20ml使溶解,滤过,滤液置分液漏斗中,加浓度为25%-28%的浓氨试液0.5ml,用三氯甲烷振摇提取2次,每次10ml,合并三氯甲烷液,蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液;另取苦参碱和槐定碱对照品,分别加无水乙醇制成每1ml含1mg的溶液,作为对照品溶液,照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取上述三种溶液各5μl,分别点于同一硅胶g薄层板上,以环己烷-丙酮-乙酸乙酯-浓氨试液=2:3:4:0.5为展开剂,置氨蒸气预饱和的展缸内,预饱和30分钟,展开,取出,晾干,喷以稀碘化铋钾试液;供试品色谱中,在于对照品色谱相应的位置上,显相同颜色的斑点;
所述黄芪的薄层鉴别方法为:取药物颗粒内容物5g,加60-90℃的石油醚30ml,超声处理20分钟,滤过,药渣挥尽石油醚,加甲醇50ml,加热回流30分钟,滤过,滤渣用少量甲醇洗涤,洗液与滤液合并,蒸干,残渣加水10ml,加热使溶解,放冷,置离心管中离心10分钟,取上清液,用三氯甲烷振摇提取2次,每次15ml,弃去三氯甲烷液,用以水饱和的正丁醇振摇提取3次,第一次加10ml,第二次加10ml,第三次加5ml,合并正丁醇液,加氨试液洗涤3次,每次15ml,弃去氨液;正丁醇液用以正丁醇饱和的水洗至中性,弃去水液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取供试品溶液10μl、对照品溶液5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水=13:7:2,10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;再置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑;
淫羊藿的薄层鉴别方法为:取药物颗粒内容物5g,加60-90℃的石油醚30ml,超声处理20分钟,滤过,药渣挥尽石油醚,加甲醇50ml,加热回流30分钟,滤过,滤渣用5ml甲醇洗涤,洗液与滤液合并,蒸干,残渣加水10ml,加热使溶解,放冷,置离心管中离心10分钟,取上清液,用三氯甲烷振摇提取2次,每次15ml,弃去三氯甲烷液,用以水饱和的正丁醇振摇提取3次,第一次加10ml,第二次加10ml,第三次加5ml,合并正丁醇液,加氨试液洗涤3次,每次15ml,弃去氨液;正丁醇液用以正丁醇饱和的水洗至中性,弃去水液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;取淫羊藿苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取供试品溶液2~4μl和对照品溶液5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水=13:7:2,10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;再置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点;
(3)含量测定:包括对苦参碱和/或淫羊藿苷的含量测定;
所述苦参碱的含量测定方法为:照高效液相色谱法(中国药典2015年版四部0512法)测定:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:甲醇为a相,0.1%三乙胺为b相,洗脱条件:53%a-47%b;检测波长为220nm;理论塔板数按苦参碱峰计算应不低于4000;
对照品溶液的制备:精密称取苦参碱对照品,加甲醇制成每1ml含0.2mg的溶液,摇匀,即得;
供试品溶液的制备:取药物颗粒内容物1.0g,精密称定,置具塞锥形瓶中,加入浓度为25%-28%的浓氨试液2ml润湿,加入三氯甲烷30ml,加热回流1.5小时,过滤,滤液蒸干,残渣加10ml甲醇溶解,摇匀,即得;
测定法:分别吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
所述淫羊藿苷的含量测定方法为:照高效液相色谱法(中国药典2010年版一部附录ⅵd)测定:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:乙腈为a相,水为b相,洗脱条件:25.5%a-74.5%b;检测波长为270nm;理论塔板数按淫羊藿苷峰计算应不低于3000;
对照品溶液的制备:取淫羊藿苷对照品,精密称定,加甲醇制成每1ml含25μg的溶液,即得;
供试品溶液的制备:取装量差异项下的药物内容物,研细,取1.5g,精密称定,置具塞锥形瓶中,精密加入70%乙醇25ml,密塞,称定重量,超声处理,1小时,超声功率250w,超声频率35khz,取出,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
发明人进行了大量的实验,以下是本发明所述检测方法的研究
实验例1:大枣薄层鉴别
1.1仪器及试药
zf-20d紫外分析仪;硅胶g板(100*100mm,批号:20100321;100*200mm;批号:20100317,厂家:青岛海洋化工厂)、硅胶h板(100*100mm,批号:20100503;100*200mm;批号:20100417,厂家:青岛海洋化工厂);硅胶h薄层板(自制)。
无水乙醇(批号:20100705;厂家:国药集团化学试剂有限公司)、乙酸乙酯(批号:1100101;厂家:上海博化有限公司)、甲醇(批号:20100901;厂家:国药集团化学试剂有限公司)、硫酸(批号:20090201;厂家:重庆川东化工有限公司)、三氯甲烷(批号:0907142;厂家:西陇科学股份有限公司)、乙醚(批号:20101125;厂家:国药集团化学试剂有限公司)、甲苯(批号:20101225;厂家:国药集团化学试剂有限公司)、冰醋酸批号:s189k040)、乙酸乙酯(批号:20090418;成都市科陇化工有限公司)。
自制薄层板:取硅胶h1份,与1%的cmc-na水溶液3份混合均匀,铺板,晾干,在105℃干燥30分钟,取出,放入干燥器中,备用。
芪胶升白胶囊(批号:2527025、2527026、2528001)、大枣阴性样品(批号:20171218)、大枣对照药材(批号:121040-201408)。对照药材购至中国药品生物制品鉴定所;正己烷、三氯甲烷、甲醇、乙醚、乙酸乙酯、冰醋酸、甲苯、乙醇等均为分析纯。
1.2薄层鉴别实验条件考察
1.2.1供试品溶液制备条件考察
条件1:取本品内容物3g,加乙酸乙酯30ml超声30分钟,滤过,滤液蒸干,残渣加甲醇0.5ml使溶解,即得。
条件2:取本品内容物3g,加乙醚30ml超声30分钟,滤过,滤液蒸干,残渣加甲醇0.5ml使溶解,即得。
条件3:取本品内容物3g,加甲醇30ml超声30分钟,滤过,滤液蒸干,残渣加甲醇0.5ml使溶解,即得。
1.2.2对照药材溶液制备
条件1:取大枣对照药材2g,加乙酸乙酯30ml,超声处理30分钟,过滤,滤液蒸干,残渣加甲醇1ml溶解,即得。
条件2:取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,加乙酸乙酯50ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,即得。
条件3:取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,加乙醚50ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,即得。
条件4:取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,加甲醇50ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,即得
1.2.3阴性供试品溶液制备:取缺大枣的阴性样品3g,照供试品溶液制法制备阴性对照溶液。
1.2.4展开及检视条件考察
条件1:照薄层色谱法(《中国药典》2015年版四部0502法[3])试验,分别取对照药材溶液10μl、供试品溶液5μl点于同一硅胶h薄层板上,分别以甲苯-乙酸乙酯-冰醋酸(60:6:0.05)、正己烷-乙酸乙酯(3:1)为展开剂,展开,取出,晾干,然后喷以10%硫酸乙醇显色剂,105℃加热至斑点清晰,再置紫外灯(365nm)下检视。
条件2:照薄层色谱法(《中国药典》2015年版四部0502法)试验,分别取对照药材溶液10μl、供试品溶液5μl点于同一硅胶g薄层板上,以甲苯-乙酸乙酯-冰醋酸(60:6:0.05)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇显色剂,105℃加热至斑点清晰,置紫外灯(365nm)下检视。
条件3:照薄层色谱法(《中国药典》2015年版四部0502法)试验,点样量考察:分别取对照药材溶液10μl、供试品溶液3μl、5μl、10μl、15μl点于同一硅胶h薄层板上,以甲苯-乙酸乙酯-冰醋酸(60:6:0.05)为展开剂,展开,取出,晾干,然后喷以10%硫酸乙醇显色剂,105℃加热至斑点清晰,再置紫外灯(365nm)下检视。
结果表明,取本品内容物3g,加乙酸乙酯、乙醚、甲醇均能提出大枣成分,从色谱图中可以看出,乙醚、甲醇对大枣的提取效果弱于乙酸乙酯,如图1:
从层析条件的色谱图中比较得出,h薄层板、甲苯-乙酸乙酯-冰醋酸(60:6:0.05)分离度好,显色清晰,如图2、图3、图4:点样量5μl为最佳条件考察图谱如图5:
结合上述实验,取本品内容物3g,加乙酸乙酯30ml,超声处理30分钟,过滤,蒸干滤液,残渣加甲醇0.5ml使溶解,作为供试品。另取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,加乙酸乙酯50ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,作为对照药材溶液,吸取供试品溶液5μl、对照药材溶液10μl,分别点于同一硅胶h薄层板上,以甲苯-乙酸乙酯-冰醋酸(60:6:0.05)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外灯(365nm)下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,斑点分离度高、显色清晰,阴性对照无干扰(如图6),故选择列入质量标准正文。
1.3耐用性
1.3.1不同湿度的比较
温度25℃时,分别在湿度35%和75%条件下进行展开。结果见(图7、8)。
1.3.2不同温度的比较
湿度75%时,分别在4℃、10℃和40℃的温度环境下进行展开。结果见(图9、10、11)。
1.3.3不同厂牌薄层板(结果见图12-图14)
实验例2苦参碱含量测定
2.1仪器与试药
岛津lc-30ad高效液相色谱仪;安捷伦1260高效液相色谱仪;kq-500vdb型双频数控超声波清洗机(昆山市超声仪器有限公司);芪胶升白胶囊有贵州汉方药业有限公司提供;苦参碱对照品(中国食品药品检定研究院,批号:110805-200508,供含量测定);试剂:甲醇(色谱纯、分析纯),乙醇(分析纯),水(wp-up-lh-40理化分析性超纯水机),三乙胺(分析纯),三氯甲烷(分析纯),正己烷(分析纯),乙醚(分析纯),磷酸(分析纯)。
2.2测定方法
1.2.1色谱条件选择:
(1)色谱柱:参照公司产品日舒安湿巾、日舒安洗液[5]中苦参碱含量测定选择十八烷基硅烷键合硅胶为填充剂的柱子进行考察。结果表明采用十八烷基硅烷键合硅胶为填充剂的柱子分离效果较好。
(2)流动相:参考2015年版药典[4]及公司产品标准,经过试验摸索,采用甲醇-0.1%三乙胺水溶液(53:47)为流动相,待测成分苦参碱及其他杂质得到较好分离。
(3)检测波长:参照2015年版药典苦参药材苦参碱含量测定,选择检测波长为220nm。
(4)理论塔板数:参照公司产品日舒安湿巾、日舒安洗液中苦参碱含量测定选择理论塔板数不少于4000。
1.2.2供试品溶液的制备:
取本品内容物1.0g,精密称定,置具塞锥形瓶中,加入浓氨试液2ml润湿,加入三氯甲烷30ml,加热回流1.5小时,过滤,滤液蒸干,残渣加10ml甲醇溶解,摇匀,即得。
1.2.3对照品溶液的制备:
精密称取苦参碱对照品适量,加甲醇制成每1ml含0.2mg的溶液,摇匀,即得。
1.3方法验证
1.3.1准确度实验
取已测定含量的芪胶升白胶囊(批号:2527025,平均含量:1.7499mg/g)约1.0g,精密称定,置具塞锥形瓶中,精密加入苦参碱对照品溶液(0.22100mg/ml)7ml,按供试品溶液制备方法制备样品溶液,并按上述色谱条件测定苦参碱的含量,计算回收率,结果见表1以及准确度色谱图(图15)进样量均为5μl,说明有较好回收率。
表1回收率试验数据(n=6)
1.3.2重复性试验
取同一批芪胶升白胶囊(2527025),按照供试品溶液制备方法制备6份,按上述色谱条件测定苦参碱的含量,结果见表2以及重复性色谱图(图16),说明有较好重复性。
表2重复性实验(n=6)
1.3.3精密度试验
精密量取同一份芪胶升白胶囊供试品溶液5μl,连续进样6次,记录色谱图,结果见表3及精密度色谱图(见图17),说明有较好精密度。
表3精密度试验(n=6)
1.3.4专属性
取芪胶升白胶囊(2527025)以及阴性样品按供试品溶液制备方法制备样品溶液以及阴性样品溶液,按上述色谱条件,分别取对照品溶液、样品溶液、阴性样品溶液5μl注入液相色谱仪,样品溶液色谱在对照品溶液色谱相应的色谱峰,且阴性无干扰,见专属性色谱图(见图18)。
1.3.5线性关系考察
按上述色谱条件,精密取苦参碱对照品(0.22100mg/ml)3μl、5μl、7μl、9μl、11μl、13μl依次注入液相色谱仪,测定峰面积。以对照品进样量x为横坐标,峰面积y为纵坐标,进行线性回归计算,绘制工作曲线(图19),得出线性回归方程为:y=48.811x+17.542,r^2=0.999。苦参碱在0.663μg-2.873μg的范围内对照品的进样量与峰面积呈良好的线性关系。见线性色谱图(图20)。
表4苦参碱对照品线性关系
1.3.6范围
按正常测定取样量的80%和120%取样测定,考察在高低取样测定的精密度。去芪胶升白胶囊(2527025)在高低取样各6份,按供试品溶液制备方法制备,按上述色谱条件测定苦参碱的含量,结果见表5。
表5范围测定结果
1.3.7耐用性
1.3.7.1样品提取溶剂的选择
取芪胶升白胶囊(2527025)样品,研细,混合均匀,取3份,每份约1.0g,精密称定,分别置具塞锥形瓶中,分别加入2ml浓氨试液润湿后,再精密加入乙醚、三氯甲烷、甲醇各30ml,加热回流1.5小时,取出,过滤,滤液蒸干,残渣加甲醇10ml溶解,得供试品溶液,分别取上述4份溶液各5μl注入液相色谱仪,计算,结果见表6,说明本品采用三氯甲烷作为提取溶剂,含量提取更完全。
表6提取溶剂的选择
1.3.7.2样品提取方法的选择
取芪胶升白胶囊(2527025)样品,研细,混合均匀,取2份每份约1.0g,精密称定,分别置具塞锥形瓶中,分别加入2ml浓氨试液润湿后精密加入三氯甲烷30ml,其中一份超声处理1小时,另一份加热回流提取1小时,取出,过滤,滤液蒸干。残渣加甲醇10ml溶解,得供试品溶液,分别取上述2份溶液各5μl注入液相色谱仪,计算,结果见表7,超声处理与回流提取含量比较,回流提取含量更完全,故选择加热回流提取。
表7提取方法的选择
1.3.7.3样品提取时间影响
取芪胶升白胶囊(2527025)样品,研细,混合均匀,取3份,,每份约1.0g,精密称定,分别置具塞锥形瓶中,分别2ml浓氨试液润湿后,再精密加入三氯甲烷30ml,分别加热回流1.0、1.5、2.0小时,取出,过滤,滤液蒸干,残渣加甲醇10ml溶解,得供试品溶液,分别取上述3份溶液各5μl注入液相色谱仪,计算,结果见表8,综合考虑,选择加热回流1.5小时为样品提取时间。
表8提取时间影响
1.3.7.4不同品牌柱子的影响
采用不同品牌十八烷基硅烷键合硅胶为填充剂的柱子,分别测定了芪胶升白胶囊(2527025)中苦参碱的含量,考察不同品牌的色谱柱的影响。色谱图见耐用性色谱图,含量测定结果见表9。结果表明不同品牌十八烷基硅烷键合硅胶为填充剂的色谱柱对含量测定无影响。
表9不同厂牌的柱子的测定结果
1.3.7.5流动相比例的影响
在不改变流动相成分条件下,改变流动相比例,考察不同比例流动相的影响,结果在所选定的流动相比例下,样品分离度较好,含量测定结果见表10和图21。
表10不同流动相比例条件下测定结果
1.3.7.6样品稳定性试验
取芪胶升白胶囊(2527025),按供试品溶液制备方法制备,按上述色谱条件测定苦参碱的含量,结果见表11和图22。
表11样品稳定性试验
1.4样品测定
按上述条件,分别测定15批样品,结果见表12。
表12样品测定结果
本品苦参药材为提取物入药,故每1g含苦参以苦参碱(c15h24n2o)计,含量限度应为:处方中苦参含量*苦参药材限度(苦参碱、氧化苦参碱总含量)/制得胶囊粒数*0.7=250g*1.2%/1000粒*0.7*1000mg=2.10mg/粒,每粒0.5g,故按每1g计算,为4.2mg/g。
由于该生产工艺过程中苦参水煮,该过程中苦参碱有部分损失,以及膏粉混合,75~95℃干燥过程中苦参碱损失严重,结合生产过程试验证明,两个过程中水煮苦参碱转移率达58%,总混干燥过程中苦参碱只能达到41.5%的转移率,总转移率只能达约40%,未能达到70%的转移率,见表13模拟苦参碱转移试验结果及苦参碱转移试验色谱图(图23)。
故暂定本品每1g含苦参碱(c15h24n2o)计,不得少于1.3mg/g。
表13模拟苦参碱转移试验结果
本发明的有益效果在于:本发明提供了一种芪胶升白胶囊的检测方法,包括以有效成分苦参碱和淫羊藿苷为指标,采用高效液相色谱法测定苦参碱和淫羊藿苷的含量;以及药物中大枣、当归、苦参、黄芪、淫羊藿的薄层色谱(tlc)鉴别方法;所述检测方法准确,灵敏度高,重复性好,检测结果稳定,增加大枣薄层色谱鉴别;测定苦参药材中苦参碱的量,为建立有效可靠的质量标准,能够准确反映中药整体质量提供数据参考。可有效控制芪胶升白胶囊的质量,既更有利于生产厂家和监督管理部门对产品质量的监测,也可以为医疗部门和患者的治疗提供更好的保障。
附图说明
图1是展开及检视条件考察结果图(1、大枣提取条件2;2大枣提取条件3;大枣提取条件;4、芪胶升白胶囊提取条件1;5、芪胶升白胶囊提取条件2;6、芪胶升白胶囊提取条件3);
图2是h板甲苯:乙酸乙酯:冰醋酸=60:6:0.05时,从层析条件的色谱图;(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图3是h板正己烷:乙酸乙酯(3:1)时,从层析条件的色谱图;(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图4是g薄层板,甲苯:乙酸乙酯:冰醋酸(60:6:0.05)时,从层析条件的色谱图;(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图5是不同点样量图谱(1、大枣对照药材溶液10μl;2、供试品3μl;3、供试品5μl;4、供试品10μl;5、供试品15μl);
图6是专属性图谱(1、大枣对照药材溶液2、25270253、25270264、25280015、缺大枣阴性样品);
图7是温度25℃、湿度35%时,耐用性结果图(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图8是温度25℃、湿度75%时,耐用性结果图(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图9是湿度75%、温度4℃时,耐用性结果图(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图10是湿度75%、温度10℃时,耐用性结果图(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图11是湿度75%、温度40℃时,耐用性结果图(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图12是湿度75%、温度25℃时,青岛海洋化工厂牌薄层板耐用性结果图(1、大枣对照药材溶液;2、2527025;3、2527026;4、2528001);
图13是湿度75%、温度25℃时,青岛海浪化工厂牌薄层板耐用性结果图;(1、大枣对照药材溶液2、25270253、25270264、2528001)
图14是湿度75%、温度25℃时,自制h薄层板耐用性结果图(1、大枣对照药材溶液2、2527025;25270264、2528001)
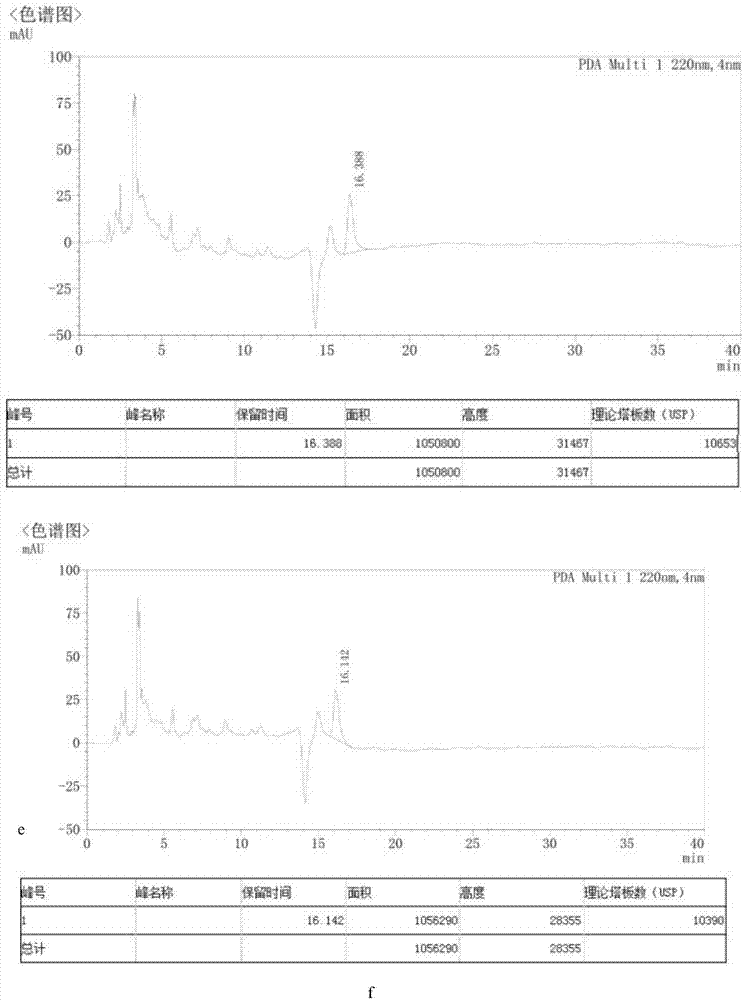
图15是苦参碱准确度色谱图;(a-苦参碱对照1,试验次数1;b-苦参碱对照2,试验次数2;c-苦参碱对照3,试验次数3;d-苦参碱对照4,试验次数4;e-苦参碱对照5,试验次数5;f-苦参碱对照6,试验次数6);
图16是苦参碱重复性色谱图(a-苦参碱对照1,试验次数1;b-苦参碱对照2,试验次数2;c-苦参碱对照3,试验次数3;d-苦参碱对照4,试验次数4;e-苦参碱对照5,试验次数5;f-苦参碱对照6,试验次数6);
图17是苦参碱精密度色谱图(a-苦参碱对照1,试验次数1;b-苦参碱对照2,试验次数2;c-苦参碱对照3,试验次数3;d-苦参碱对照4,试验次数4;e-苦参碱对照5,试验次数5;f-苦参碱对照6,试验次数6);
图18是苦参碱专属性色谱图(a-苦参碱对照1,试验次数1;b-苦参碱对照2,试验次数2;c-苦参碱对照3);
图19是苦参碱含量测定时,线性回归曲线图;
图20是苦参碱含量测定时,线性色谱图(a-苦参碱对照1,试验次数1;b-苦参碱对照2,试验次数2;c-苦参碱对照3,试验次数3;d-苦参碱对照4,试验次数4;e-苦参碱对照5,试验次数5;f-苦参碱对照6,试验次数6);
图21是苦参碱含量测定时,流动相比例影响结果图;(a-甲醇:0.1%三乙胺水溶液=53:47;b-甲醇:0.1%三乙胺水溶液=50:45;c-甲醇:0.1%三乙胺水溶液=50:50);
图22是苦参碱含量测定时,稳定性色谱图;
图23是苦参碱含量测定时,苦参碱工艺模拟色谱图;其中a是工艺模拟序列1(苦参碱对照品);b是工艺模拟序列2(25278001总混前浸膏);c是工艺模拟序列3(25278001总混后);d是工艺模拟序列4(25278002总混前浸膏);e是工艺模拟序列5(25278002总混后);f是工艺模拟序列6(苦参碱、氧化苦参碱混合对照);g是工艺模拟序列7(苦参药材zy17112001);h是工艺模拟序列8(苦参药材zy17112002)。
下面结合实施例对本发明作进一步的说明
具体实施方式
实施例1:芪胶升白胶囊的检测方法为
配方:大枣250g、阿胶250g、血人参375g、淫羊藿375g、苦参250g、黄芪750g、当归250g。
工艺:以上七味,除阿胶外,当归粉碎成细粉;其余大枣等五味,加水煎煮三次,第一次2小时,第二次1.5小时,第三次1小时,合并煎液,滤过,滤液加入烊化后的阿胶,搅匀,浓缩至75-85℃下相对密度为1.30的稠膏,加入上述当归细粉,混匀,制成颗粒,干燥,装入胶囊,即得。
检测方法:
(1)性状:硬胶囊内容物为棕褐色的颗粒及粉末;味苦;
(2)鉴别:包括对大枣的薄层鉴别、当归的薄层鉴别、苦参的薄层鉴别、黄芪的薄层鉴别和/或淫羊藿的薄层鉴别;
所述大枣的薄层鉴别为:取药物颗粒内容物3g,加乙酸乙酯30ml,超声处理30分钟,过滤,蒸干滤液,残渣加甲醇0.5ml使溶解,作为供试品溶液;另取大枣对照药材2g,加水50ml,加热回流2小时,趁热过滤,滤液挥干水分制成大枣稠膏,膏加乙酸乙酯30ml,超声处理30分钟,过滤,滤液蒸干,加甲醇1ml使溶解,作为对照药材溶液,吸取供试品溶液5μl和对照药材溶液10μl,分别点于同一硅胶h薄层板上,以甲苯-乙酸乙酯-冰醋酸=60:6:0.05为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点;
所述当归的薄层鉴别方法为:取药物颗粒内容物5g,加正己烷20ml,超声处理20分钟,滤过,滤液挥至1ml,作为供试品溶液;另取当归对照药材0.5g,同法制成对照药材溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取上述两种溶液各5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯=9:1为展开剂,展开,取出,晾干,置波长为365nm紫外灯下检视,供试品色谱中,在于对照药材色谱相应的位置上,先相同颜色的荧光斑点;
所述苦参的薄层鉴别方法为:取药物颗粒内容物1g,加乙醇30ml,加热回流30分钟,放冷,滤过,滤液蒸干残渣加水20ml使溶解,滤过,滤液置分液漏斗中,加浓度为25%-28%的浓氨试液0.5ml,用三氯甲烷振摇提取2次,每次10ml,合并三氯甲烷液,蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液;另取苦参碱和槐定碱对照品,分别加无水乙醇制成每1ml含1mg的溶液,作为对照品溶液,照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取上述三种溶液各5μl,分别点于同一硅胶g薄层板上,以环己烷-丙酮-乙酸乙酯-浓氨试液=2:3:4:0.5为展开剂,置氨蒸气预饱和的展缸内,预饱和30分钟,展开,取出,晾干,喷以稀碘化铋钾试液;供试品色谱中,在于对照品色谱相应的位置上,显相同颜色的斑点;
所述黄芪的薄层鉴别方法为:取药物颗粒内容物5g,加60-90℃的石油醚30ml,超声处理20分钟,滤过,药渣挥尽石油醚,加甲醇50ml,加热回流30分钟,滤过,滤渣用少量甲醇洗涤,洗液与滤液合并,蒸干,残渣加水10ml,加热使溶解,放冷,置离心管中离心10分钟,取上清液,用三氯甲烷振摇提取2次,每次15ml,弃去三氯甲烷液,用以水饱和的正丁醇振摇提取3次,第一次加10ml,第二次加10ml,第三次加5ml,合并正丁醇液,加氨试液洗涤3次,每次15ml,弃去氨液;正丁醇液用以正丁醇饱和的水洗至中性,弃去水液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取供试品溶液10μl、对照品溶液5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水=13:7:2,10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;再置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑;
淫羊藿的薄层鉴别方法为:取药物颗粒内容物5g,加60-90℃的石油醚30ml,超声处理20分钟,滤过,药渣挥尽石油醚,加甲醇50ml,加热回流30分钟,滤过,滤渣用5ml甲醇洗涤,洗液与滤液合并,蒸干,残渣加水10ml,加热使溶解,放冷,置离心管中离心10分钟,取上清液,用三氯甲烷振摇提取2次,每次15ml,弃去三氯甲烷液,用以水饱和的正丁醇振摇提取3次,第一次加10ml,第二次加10ml,第三次加5ml,合并正丁醇液,加氨试液洗涤3次,每次15ml,弃去氨液;正丁醇液用以正丁醇饱和的水洗至中性,弃去水液,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;取淫羊藿苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液;照薄层色谱法(中国药典2010年版一部附录ⅵb)试验,吸取供试品溶液2~4μl和对照品溶液5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水=13:7:2,10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;再置波长为365nm紫外灯下检视,在供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点;
(3)含量测定:包括对苦参碱和/或淫羊藿苷的含量测定;
所述苦参碱的含量测定方法为:照高效液相色谱法(中国药典2015年版四部0512法)测定:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:甲醇为a相,0.1%三乙胺为b相,洗脱条件:53%a-47%b;检测波长为220nm;理论塔板数按苦参碱峰计算应不低于4000;
对照品溶液的制备:精密称取苦参碱对照品,加甲醇制成每1ml含0.2mg的溶液,摇匀,即得;
供试品溶液的制备:取药物颗粒内容物1.0g,精密称定,置具塞锥形瓶中,加入浓度为25%-28%的浓氨试液2ml润湿,加入三氯甲烷30ml,加热回流1.5小时,过滤,滤液蒸干,残渣加10ml甲醇溶解,摇匀,即得;
测定法:分别吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
所述淫羊藿苷的含量测定方法为:照高效液相色谱法(中国药典2010年版一部附录ⅵd)测定:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:乙腈为a相,水为b相,洗脱条件:25.5%a-74.5%b;检测波长为270nm;理论塔板数按淫羊藿苷峰计算应不低于3000;
对照品溶液的制备:取淫羊藿苷对照品,精密称定,加甲醇制成每1ml含25μg的溶液,即得;
供试品溶液的制备:取装量差异项下的药物内容物,研细,取1.5g,精密称定,置具塞锥形瓶中,精密加入70%乙醇25ml,密塞,称定重量,超声处理,1小时,超声功率250w,超声频率35khz,取出,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
- 还没有人留言评论。精彩留言会获得点赞!