一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法与流程
本发明涉及分析化学领域,尤其涉及一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法。
背景技术:
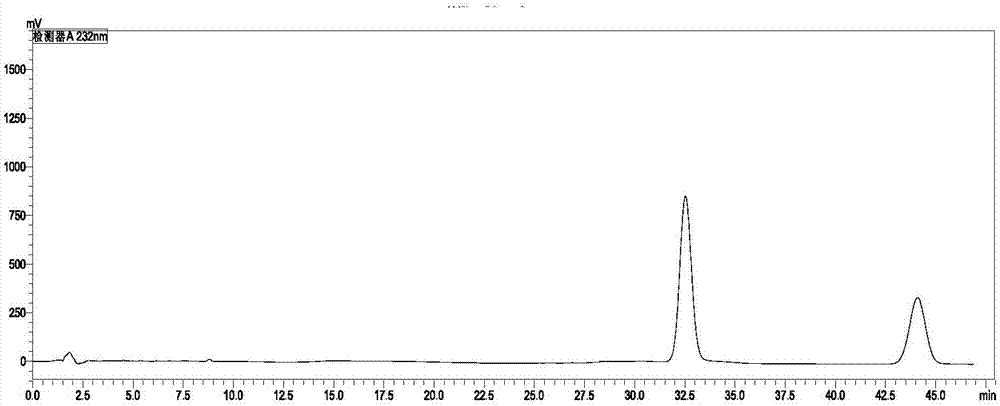
氰氟草酯和嘧啶肟草醚复配乳油或复配油悬剂,是一种复合型除草剂,对禾本科杂草除草效果显著。目前国内外氰氟草酯和嘧啶肟草醚的含量一起测定方法未查到相关专利,而氰氟草酯成分检测方法是高效液相色谱正向检测方法,嘧啶肟草醚成分检测方法是高效液相色谱反相检测方法,且目前公开的方法单独检测每一个成分耗时长并且浪费试剂,并且照搬检测方法杂质峰与有效成分峰分离不开。所以,为了氰氟草酯和嘧啶肟草醚复配制剂使用的发展,提供一种检测氰氟草酯和嘧啶肟草醚的复配制剂中氰氟草酯和嘧啶肟草醚含量的方法具有重要的意义。技术实现要素:有鉴于此,本发明所要解决的技术问题在于提供一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法,本发明提供的方法无需对待测的复配试剂进行处理,且检测结果准确,灵敏度高,重现性好。本发明提供了一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法,包括:将氰氟草酯和嘧啶肟草醚的复配制剂待测样通过高效液相色谱进行检测,得到氰氟草酯的含量和嘧啶肟草醚的含量;其中,所述高效液相色谱检测的波长为225~240nm;所述高效液相色谱检测的流动相为乙腈和水的混合物,且所述乙腈和水的体积比为(58~62)∶(38~42)。优选的,所述高效液相色谱检测的色谱柱为键合固定相色谱柱。优选的,所述键合固定相色谱柱为c30、c18、c8或c4柱。优选的,所述色谱柱的柱长为1~1000mm。优选的,所述色谱柱的柱内径为0.1~50mm。优选的,所述乙腈和水的体积比优选为58∶42、59∶41、60∶40、61∶39或62∶38。优选的,所述流动相的流速为0.8~1.2ml/min。优选的,所述流动相的流速为1ml/min。优选的,所述氰氟草酯和嘧啶肟草醚的复配制剂待测样按照以下方法制备得到:将氰氟草酯和嘧啶肟草醚的复配制剂和乙腈混合,得到氰氟草酯和嘧啶肟草醚的复配制剂待测样。优选的,所述氰氟草酯和嘧啶肟草醚的复配制剂为氰氟草酯和嘧啶肟草醚复配乳油制剂或氰氟草酯和嘧啶肟草醚复配油悬剂。与现有技术相比,本发明提供了一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法,包括:将氰氟草酯和嘧啶肟草醚的复配制剂待测样通过高效液相色谱进行检测,得到氰氟草酯的含量和嘧啶肟草醚的含量;其中,本发明通过选择所述高效液相色谱检测的波长为225~240nm;所述高效液相色谱检测的流动相为乙腈和水的混合物,且所述乙腈和水的体积比为(58~62)∶(38~42),进而使得本发明提供的方法无需对种氰氟草酯和嘧啶肟草醚的复配制剂进行预处理即可进行检测,且检测结果准确,重现性好,灵敏度高。附图说明图1为氰氟草酯和嘧啶肟草醚标准品溶液的色谱图;图2为氰氟草酯和嘧啶肟草醚乳油待测样品的色谱图;图3为氰氟草酯和嘧啶肟草醚乳油待测样品在流动相乙腈与水的体积比为55∶45时得到的色谱图;图4为氰氟草酯和嘧啶肟草醚乳油待测样品在流动相乙腈与水的体积比为65∶35时得到的色谱图;图5为氰氟草酯和嘧啶肟草醚乳油待测样品在流动相乙腈与水的体积比为70∶30时得到的色谱图。具体实施方式本发明提供了一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法,包括:将氰氟草酯和嘧啶肟草醚的复配制剂待测样通过高效液相色谱进行检测,得到氰氟草酯和嘧啶肟草醚的含量;其中,所述高效液相色谱检测的波长为225~240m;所述高效液相色谱检测的流动相为乙腈和水的混合物,且所述乙腈和水的体积比为(58~62)∶(38~42)。本发明中,所述高效液相色谱检测的色谱柱优选为键合固定相色谱柱,更优选为为c30、c18、c8或c4柱;其中,所述色谱柱的柱内径优选为0.1~50mm,更优选为1~10mm,更优选为4.6~6mm;所述色谱柱的柱长优选为1~1000mm,更优选为200~800mm,最优选为232~500mm;所述色谱柱填料粒度优选为0.1~20μm,更优选为5.0~10μm。本发明中,所述高效液相色谱检测的检测器优选为可变波长紫外检测器;所述高效液相色谱检测的波长优选为225~240nm,更优选为232nm。本发明中,所述乙腈和水的体积比优选为58∶42、59∶41、60∶40、61∶39或62∶38,更优选为60∶40。所述流动相的流速优选为0.8~1.2ml/min,更优选为1ml/min。本发明中,所述氰氟草酯和嘧啶肟草醚的复配制剂待测样按照以下方法制备得到:将氰氟草酯和嘧啶肟草醚的复配制剂和乙腈混合,得到氰氟草酯和嘧啶肟草醚的复配制剂待测样。其中,本发明对复配制剂的剂型并没有特殊要求,如可以为氰氟草酯和嘧啶肟草醚复配乳油制剂或氰氟草酯和嘧啶肟草醚复配油悬剂。本发明中,所述待测样的进样量优选为5μl。对于待测样的计算方法优选采用外标法进行计算得到。本发明提供的一种检测复配制剂中氰氟草酯和嘧啶肟草醚含量的方法通过将氰氟草酯和嘧啶肟草醚的复配制剂待测样通过高效液相色谱进行检测,得到复配制剂中氰氟草酯和嘧啶肟草醚的含量;其中,本发明通过选择所述高效液相色谱检测的波长为225~240nm;所述高效液相色谱检测的流动相为乙腈和水的混合物,且所述乙腈和水的体积比为(58~62)∶(38~42),进而使得本发明提供的方法无需对种氰氟草酯和嘧啶肟草醚的复配制剂进行预处理排出干扰即可进行检测,大大简化了高效液相色谱的检测过程,且检测结果准确,重现性好,灵敏度高。下面将结合本发明实施例的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。各实施例所用仪器均为岛津lc-20a高效液相色谱仪;氰氟草酯和嘧啶肟草醚标准品为fluka标准品。实施例1检测的色谱条件如下:可变波长紫外检测器,c18色谱柱5.0μm250mm×4.6mm(i.d.);流动相:乙腈∶水=60∶40;检测波长:232nm;流速:1.0ml/min;具体检测方法为:准确称取标准品氰氟草酯0.05g和嘧啶肟草醚0.0210g置于同一50ml容量瓶中,用乙腈溶解并稀释至刻度。称取60g/l氰氟草酯+25g/l嘧啶肟草醚复配乳油的样品0.8100g左右置于50ml容量瓶中,用乙腈溶解并稀释至刻度,超声波脱气5分钟。取标样进行液相色谱分析,进样量为5μl,测定氰氟草酯和嘧啶肟草醚的峰面积。取样品进行液相色谱分析,进样量为5μl,进行检测。氰氟草酯和嘧啶肟草醚标准品溶液的色谱图如图1所示,图1为氰氟草酯和嘧啶肟草醚标准品溶液的色谱图,从图中可以看出,32.5min为氰氟草酯的峰和44.1min为嘧啶肟草醚的峰。氰氟草酯和嘧啶肟草醚乳油待测样品谱图见图2,图2为氰氟草酯和嘧啶肟草醚乳油待测样品的色谱图,从图中可以看出,主峰的出峰时间适宜,且与杂峰分离度良好,用外标法分别计算得出样品中氰氟草酯和嘧啶肟草醚含量为5.67%、2.36%。将上述样品溶液中加入标准品氰氟草酯和嘧啶肟草醚,按上述实验条件,对该样品作四个加标含量,每个加标含量作3次平行测定取平均值,结果见表1,根据实际加入量和实测结果,计算该样品的加标回收率在99%~101%之间。表1实施例2为验证实施例1检测方法的可行性和准确性,进行精密度试验,试验采用浓度为1000mg/l配制的氰氟草酯和嘧啶肟草醚标准溶液,取标准溶液5μl连续进样6次,测定结果表2所示。表2序号氰氟草酯峰面积嘧啶肟草醚峰面积135305737195774922353785621962941533530945619584366435287942195079635354329531945993763540768919566193平均值3535372319554228rsd%0.160.28由表2可以得出峰面积的rsd小于2%,因此此方法稳定性良好。实施例3为了更好的说明本发明实施例提供的含有氰氟草酯和嘧啶肟草醚样品溶液的稳定性,将样品溶液配制好后分别放置4h、8h、12h、24h再进行也液相色谱分析,其结果如表3所示。表3由表3可知出峰面积的rsd小于2%,本发明提供的供试品溶液稳定性良好。对比例1按照实施例1的方法对60g/l氰氟草酯+25g/l嘧啶肟草醚复配乳油进行检测,仅改变流动相乙腈和水的体积比,其它检测条件和检测方法均不变,结果如下:流动相:乙腈∶水的体积比为55∶45时,得到的谱图见图3,图3为氰氟草酯和嘧啶肟草醚乳油待测样品在流动相乙腈与水的体积比为55∶45时得到的色谱图,从图中可以看出,保留时间71.8min的峰为氰氟草酯,保留时间110.08min的峰为嘧啶肟草醚,可以看出在此流动相条件下检测耗时增加了一倍。流动相:乙腈∶水的体积比为65∶35时,得到的谱图见图4,图4为氰氟草酯和嘧啶肟草醚乳油待测样品在流动相乙腈与水的体积比为65∶35时得到的色谱图,从图中可以看出,保留时间22.5min的峰为氰氟草酯,保留时间26.5min的峰为嘧啶肟草醚,而嘧啶肟草醚的峰和杂质峰完全重合,在此流动相下不能分离出目标峰。流动相乙腈:水的体积比为70∶30时,得到的谱图见图5,图5为氰氟草酯和嘧啶肟草醚乳油待测样品在流动相乙腈与水的体积比为70∶30时得到的色谱图,从图中可以看出,在此流动相下氰氟草酯的保留时间为15.2min,嘧啶肟草醚的保留时间为19.3min,从谱图中可以看出目标峰完全和杂质峰重合,在此流动相下完全不能分离出目标峰。以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1