卡马西平的高效液相检测方法与流程
本发明涉及药物分析技术领域,具体涉及一种卡马西平的高效液相检测方法。
背景技术:
卡马西平(carbamazepine),是一类抗癫痫、稳定情绪类药物,对大发作、局限性发作和混合型癫痫亦有疗效。主要用于治疗癫痫症、躁郁症以及三叉神经痛。卡马西平具有很多副作用,其中包括:能危及生命的过敏反应,对三叉神经有毒,可能导致皮肤及内脏器官严重伤害,因此在治疗期间对病人的血液药物水平进行监测非常重要。
目前,检测血浆中卡门西平血药浓度的常用方法有:紫外分光光度法、荧光偏振免疫法、高效液相色谱法、气相色谱-质谱联用法、液相色谱-质谱联用法,这些方法中,液相色谱法为卡门西平的首选方法。以往检测方法存在前处理复杂、干扰因素多、分析时间长、灵敏度不高、定性定量不够准确的问题。
中国药典2015年版,记载了卡马西平的有关物质的检测方法,其色谱条件为:以色谱条件与系统适用性试验腈丙基硅烷键合硅胶为填充剂,以甲醇-四氢呋喃-水(120:30:850)为流动相(加入0.02%甲酸;0.05%三乙胺),检测波长为230nm,20ul进样。另,现有技术已报道卡马西平的含量测定方法,亦采用了腈丙基硅烷键合硅胶为填充剂的色谱柱,流动相a为50mmol/l磷酸二氢铵溶液,流动相b采用乙腈,梯度洗脱时间65min,进样50ul,在215nm检测。发明人按该方法对卡马西平含量及其杂质进行测定,发现hplc图谱洗脱时基线有漂移,积分不准确,其用于定量分析时的精确度和准确度不高,且检测需要65min以上,耗时长。
技术实现要素:
本发明所要解决的技术问题是,提供一种检测时间短、分离效果好、精确度和准确度更高的卡马西平的高效液相检测方法。
为解决上述技术问题,本发明提供的卡马西平的高效液相检测方法,其中,
色谱柱采用反相c18色谱柱;
检测器采用dad检测器;
流动相包括流动相a和流动相b;流动相a是甲酸-乙酸铵的水溶液和乙腈的混合溶液,水溶液中甲酸体积浓度为0.01~0.03%,水溶液中乙酸铵浓度为0.5~1.5mm,水溶液与乙腈的体积比为100~95:0~5;流动相b是甲酸-乙酸铵的乙腈溶液和水的混合溶液,乙腈溶液中甲酸体积浓度为0.01~0.03%,乙腈溶液中乙酸铵浓度为0.5~1.5mm,乙腈溶液与水的体积比为100~95:0~5;
采用梯度洗脱,梯度洗脱程序按以下程序进行:0~0.01min,流动相b保持体积百分比为20%;0.01~2.10min,流动相b体积百分比由20%递增至90%;2.10~2.11min,流动相b体积百分数由90%递减至20%;2.11~3.00min,流动相b保持体积百分数为20%。
在一个优选的实施例中,流动相a是甲酸—乙酸铵的水溶液和乙腈的体积分数为95:5的混合溶液,流动相b是甲酸—乙酸铵的乙腈溶液和水的体积分数为95:5的混合溶液。
在一个优选的实施例中,流动相a中,水溶液中甲酸体积浓度为0.025%;流动相b中,乙腈溶液中甲酸体积浓度为0.025%。
在一个优选的实施例中,流动相a中,水溶液中乙酸铵浓度为1mm;流动相b中,乙腈溶液中乙酸铵浓度为1mm。
在一个优选的实施例中,色谱柱柱长为100mm。
在一个优选的实施例中,流速为1.0~2.0ml/min,更优选为1.4ml/min。
在一个优选的实施例中,检测波长为280~285nm,更优选为284nm。
在一个优选的实施例中,柱温控制在35~40℃,更优选为40℃。
在一个优选的实施例中,进样量为2~8μl,更优选为3~5μl。
本发明还提供一种测定卡马西平含量的高效液相方法,包括以下步骤:
(1)对照品溶液及供试品溶液的制备:精密称取适量卡马西平对照品,用二亚甲基亚砜溶解并定容成具有一定浓度梯度的多个对照品溶液;精密称取适量卡马西平供试品,用二亚甲基亚砜溶解并定容得到供试品溶液;
(2)色谱条件:
色谱柱采用反相c18色谱柱;
检测器采用dad检测器;
流动相包括流动相a和流动相b;流动相a是甲酸—乙酸铵的水溶液和乙腈的混合溶液,水溶液中甲酸体积浓度为0.025%,水溶液中乙酸铵浓度为1mm,水溶液与乙腈的体积比为95:5;流动相b是甲酸—乙酸铵的乙腈溶液和水的混合溶液,乙腈溶液中甲酸体积浓度为0.025%,乙腈溶液中乙酸铵浓度为1mm,乙腈溶液与水的体积比为95:5;
采用梯度洗脱,梯度洗脱程序按以下程序进行:0~0.01min,流动相b保持体积百分比为20%;0.01~2.10min,流动相b体积百分比由20%递增至90%;2.10~2.11min,流动相b体积百分数由90%递减至20%;2.11~3.00min,流动相b保持体积百分数为20%;
(3)测定方法:
将所述多个对照品溶液以及供试品溶液按所述(2)色谱条件依次进样,记录色谱图,根据多个对照品溶液的图谱数据和浓度数据制备线性相关工作曲线,代入供试样品的图谱数据计算并得出供试品溶液浓度,根据得到的供试品溶液浓度和卡马西平供试品的称取量计算卡马西平含量,完成卡马西平含量的测定。
在一个优选的实施例中,对照品溶液的浓度依次为6.25、12.5、25、50、100、200μg/ml,流速为1.4ml/min。检测波长为284nm,柱温为40℃,进样量为3~5μl。
本发明提供的卡马西平的高效液相检测方法的技术优势在于:
1.本发明的方法能够有效检出卡马西平及其有关物质,并且分离度r能达1.5以上。
2.本发明的方法测定hplc谱基线平稳,不发生漂移。
3.本发明的方法检测时间短,只需3min即可完成高效液相检测过程,本方法大大提高了检测效率,适合高通量筛选。
4.本发明的方法能节省溶剂,降低成本,操作安全简易,处理方便快捷。
5.本发明的方法用于卡马西平的含量测定,具有良好的线性关系,重现性高,在原料药、制剂质量研究以及相关药动学定量研究等方面具有重要研究价值。
附图说明
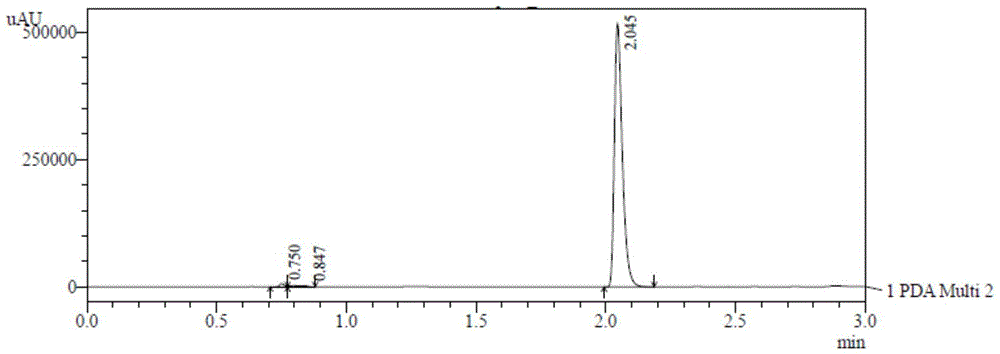
图1为采用本发明的方法测定卡马西平供试品的hplc图谱。
图2为本发明实施例1测定卡马西平供试品的hplc图谱一。
图3为本发明实施例1测定卡马西平供试品的hplc图谱二。
图4为本发明实施例1测定卡马西平供试品的hplc图谱三。
图5为本发明实施例1测定卡马西平供试品的hplc图谱四。
图6为采用本发明的方法绘制的卡马西平工作曲线。
具体实施方式
本申请的发明人经过广泛的研究和大量的实验,摸索得到了一种卡马西平的高效液相检测方法,该方法提供了能够有效分离卡马西平及其有关物质的流动相体系及梯度洗脱程序,可以将现有技术中难以有效分离的卡马西平及其有关物质实现很好的分离效果,分离度r可达1.5以上。
发明人采用的卡马西平的高效液相检测方法是:
色谱柱采用反相c18色谱柱,比如以十八烷基硅烷键合硅胶为填充剂的watersxbridge色谱柱;
检测器采用dad检测器;
流动相包括流动相a和流动相b;流动相a是甲酸—乙酸铵的水溶液/乙腈(100~95/0~5;v/v)的混合溶液,水溶液中甲酸体积浓度为0.01~0.03%,水溶液中乙酸铵浓度为0.5~1.5mm;流动相b是甲酸—乙酸铵的乙腈溶液/水(100~95/0~5;v/v)的混合溶液,乙腈溶液中甲酸体积浓度为0.01~0.03%,乙腈溶液中乙酸铵浓度为0.5~1.5mm;
采用梯度洗脱,流动相a+流动相b=100%,梯度洗脱程序按以下程序进行:0~0.01min,流动相b保持体积百分比为20%;0.01~2.10min,流动相b体积百分比由20%递增至90%;2.10~2.11min,流动相b体积百分数由90%递减至20%;2.11~3.00min,流动相b保持体积百分数为20%。
本发明的一个优选的实施例中,流动相a是甲酸—乙酸铵的水溶液和乙腈的体积分数为95:5的混合溶液,流动相b是甲酸—乙酸铵的乙腈溶液和水的体积分数为95:5的混合溶液。
本发明的一个优选的实施例中,流动相a中,水溶液中甲酸体积浓度为0.025%;流动相b中,乙腈溶液中甲酸体积浓度为0.025%。
本发明的一个优选的实施例中,流动相a中,水溶液中乙酸铵浓度为1mm;流动相b中,乙腈溶液中乙酸铵浓度为1mm。
本发明的一个优选的实施例中,色谱柱柱长为100mm。
本发明的一个优选的实施例中,流速为1.0~2.0ml/min,更优选为1.4ml/min。
本发明的一个优选的实施例中,检测波长为280~285nm,更优选为284nm。
本发明的一个优选的实施例中,柱温控制在35~40℃,更优选为40℃。
本发明的一个优选的实施例中,进样量为2~8μl,更优选为3~5μl。
下面将结合具体实施例对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例中的实验条件如下:
仪器:高效液相色谱仪(lc-20a)
色谱柱:watersxbridgec18色谱柱,柱长为100mm;
供试品溶液:精密称取卡马西平供试品10mg,置于10ml容量瓶,用二甲基亚砜溶解并定容,得到供试品溶液;
流速:1.4ml/min;
检测波长:284nm;
柱温:40℃;
流动相:流动相a+流动相b=100%;
梯度洗脱:0~0.01min,流动相b保持体积百分比为20%;0.01~2.10min,流动相b体积百分比由20%递增至90%;2.10~2.11min,流动相b体积百分数由90%递减至20%;2.11~3.00min,流动相b保持体积百分数为20%;
实施例1
流动相a是甲酸—乙酸铵的水溶液/乙腈(95/5;v/v)的混合溶液,水溶液中甲酸体积浓度为0.025%,水溶液中乙酸铵浓度为1mm;流动相b是甲酸—乙酸铵的乙腈溶液/水(95/5;v/v)的混合溶液,乙腈溶液中甲酸体积浓度为0.025%,乙腈溶液中乙酸铵浓度为1mm。
此方法下经过四次进样,hplc图谱如图2-5所示,基线平稳,重复性好,3min能够全部出峰,卡马西平保留时间为2.043~2.045min,工艺杂质保留时间为0.750~0.847,且分离度均大于1.5,理论塔板数为21509。
实施例2
流动相a是甲酸—乙酸铵的水溶液/乙腈(95/5;v/v)的混合溶液,水溶液中甲酸体积浓度为0.01%,水溶液中乙酸铵浓度为0.5mm;流动相b是甲酸—乙酸铵的乙腈溶液/水(95/5;v/v)的混合溶液,乙腈溶液中甲酸体积浓度为0.01%,乙腈溶液中乙酸铵浓度为0.5mm。
此方法下基线平稳,卡马西平保留时间为2.045min左右,且分离度为13.505。
实施例3
流动相a是甲酸—乙酸铵的水溶液/乙腈(95/5;v/v)的混合溶液,水溶液中甲酸体积浓度为0.03%,水溶液中乙酸铵浓度为1.5mm;流动相b是甲酸—乙酸铵的乙腈溶液/水(95/5;v/v)的混合溶液,乙腈溶液中甲酸体积浓度为0.03%,乙腈溶液中乙酸铵浓度为1.5mm。
此方法下基线平稳,卡马西平保留时间为2.044min左右,且分离度为13.443。
实施例4
流动相a是甲酸—乙酸铵的水溶液/乙腈(95/5;v/v)的混合溶液,水溶液中甲酸体积浓度为0.025%,水溶液中乙酸铵浓度为1mm;流动相b是甲酸-乙酸铵的乙腈溶液,乙腈溶液中甲酸体积浓度为0.025%,乙腈溶液中乙酸铵浓度为1mm。
此方法下基线平稳,卡马西平保留时间为2.043min左右,且分离度为13.801。
实施例5
流动相a是甲酸-乙酸铵的水溶液,水溶液中甲酸体积浓度为0.025%,水溶液中乙酸铵浓度为1mm;流动相b是甲酸-乙酸铵的乙腈溶液,乙腈溶液中甲酸体积浓度为0.025%,乙腈溶液中乙酸铵浓度为1mm。
此方法下基线平稳,卡马西平保留时间为2.043min左右,且分离度为13.892。
实施例6
流动相a是甲酸-乙酸铵的水溶液,水溶液中甲酸体积浓度为0.025%,水溶液中乙酸铵浓度为1mm;流动相b是甲酸-乙酸铵的乙腈溶液,乙腈溶液中甲酸体积浓度为0.025%,乙腈溶液中乙酸铵浓度为1mm。
流动相:流动相a+流动相b=100%;
本实施例的梯度洗脱调整为:0~0.01min,流动相b保持体积百分比为10%;0.01~2.10min,流动相b体积百分比由10%递增至80%;2.10~2.11min,流动相b体积百分数由80%递减至10%;2.11~3.00min,流动相b保持体积百分数为10%;
此方法下出现基线不平稳、分离度小于1、保留时间过长。
基于上述的检测方法,发明人还提供一种测定卡马西平含量的高效液相方法,按以下步骤进行:
(1)对照品溶液及供试品溶液的制备:精密称取卡马西平对照品,用二甲基亚砜溶解并定容得到1mg/ml的储备液,准确吸取50μl储备液,用二甲基亚砜依次稀释定容得到6.25、12.5、25、50、100、200ug/ml的对照品溶液;精密称取卡马西平供试品,用二甲基亚砜溶解并定容得到供试品溶液;
(2)色谱条件:
色谱柱采用反相c18色谱柱;
检测器采用dad检测器;
流动相包括流动相a和流动相b;流动相a是甲酸—乙酸铵的水溶液/乙腈(95/5;v/v)的混合溶液,水溶液中甲酸体积浓度为0.025%,水溶液中乙酸铵浓度为1mm;流动相b是甲酸—乙酸铵的乙腈溶液/水(95/5;v/v)的混合溶液,乙腈溶液中甲酸体积浓度为0.025%,乙腈溶液中乙酸铵浓度为1mm;
采用梯度洗脱,梯度洗脱程序按以下程序进行:0~0.01min,流动相b保持体积百分比为20%;0.01~2.10min,流动相b体积百分比由20%递增至90%;2.10~2.11min,流动相b体积百分数由90%递减至20%;2.11~3.00min,流动相b保持体积百分数为20%;
(3)测定方法:
将所述6个对照品溶液和供试品溶液按所述色谱条件依次进样,记录色谱图,根据6个对照品溶液的卡马西平峰面积和浓度数据制备线性相关工作曲线(如图6所示),该工作曲线线性关系良好,r≥0.9999;代入供试样品的卡马西平峰面积计算并得出供试品溶液浓度,比较测定的供试品溶液浓度和定容的供试品溶液浓度以及进样时的稀释倍数,计算得出卡马西平的含量。
本发明的卡马西平的高效液相检测方法,能够有效检出卡马西平及其有关物质,并且分离度r能达1.5以上,而现有技术中的检测方法难以实现卡马西平及其有关物质的有效分离;本发明的检测方法的hplc谱基线稳定,不发生漂移;本发明的方法检测时间短,只需3min即可完成高效液相检测过程,而现有技术中的检测方法则需要至少65min以上才能完成检测,本方法大大提高了检测效率,适合高通量筛选;本发明的方法能快速完成分析且节省溶剂,降低成本,操作安全简易,处理方便快捷;本发明的方法用于卡马西平的含量测定,具有良好的线性关系,r≥0.9999,重现性高,在原料药、制剂质量研究以及相关药动学定量研究等方面具有重要研究价值。
综上所述,上述各实施例仅为本发明的较佳实施例而已,并不用以限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,皆应包含在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!