一种(S)-(+)-3-羟基四氢呋喃对映异构体含量检测方法与流程
本发明涉及一种(s)-(+)-3-羟基四氢呋喃对映异构体含量的检测方法,属于原料药质量控制技术领域。
背景技术:
(s)-(+)-3-羟基四氢呋喃((s)-(+)-3-hydroxytetrahydrofuran),其结构式如下:
技术实现要素:
(s)-(+)-3-羟基四氢呋喃为一种药用中间体,可作为制备恩格列净、阿法替尼、安普那韦等原料药的原料。
由于(s)-(+)-3-羟基四氢呋喃的对映异构杂质会影响(s)-(+)-3-羟基四氢呋喃含量造成反应投料比的差异,故需要检测其对映异构体(r)-(+)-3-羟基四氢呋喃含量,控制(s)-(+)-3-羟基四氢呋喃质量。
紫外扫描发现(s)-(+)-3-羟基四氢呋喃仅末端波长193nm处有吸收,因此,不能采用高效液相色谱法检测(s)-(+)-3-羟基四氢呋喃中的对映异构体杂质(r)-(+)-3-羟基四氢呋喃。目前没有检测(s)-(+)-3-羟基四氢呋喃异构体的文献公开。(s)-(+)-3-羟基四氢呋喃沸点为181℃,可以使用气相色谱检测,试验证明cp-chirasildexcb气相色谱柱(规格:25m×0.25mm×0.25µm)、supelcogammadextm225气相色谱柱(规格:30m×0.25mm×0.25µm)和cyclosil-b气相色谱柱(规格:30m×0.25mm×0.25µm)均无法分离(s)-(+)-3-羟基四氢呋喃及其有对映异构体杂质(r)-(+)-3-羟基四氢呋喃,因而,无法对(s)-(+)-3-羟基四氢呋喃中的对映异构体杂质(r)-(+)-3-羟基四氢呋喃进行检测。
发明内容
发明目的:本发明的目的在于提供一种(s)-(+)-3-羟基四氢呋喃中对映异构体杂质(r)-(+)-3-羟基四氢呋喃的检测方法。
技术方案
试验发现,在检测(s)-(+)-3-羟基四氢呋喃中对映异构体杂质(r)-(+)-3-羟基四氢呋喃含量前,对(s)-(+)-3-羟基四氢呋喃进行衍生化处理,将样品中的(s)-(+)-3-羟基四氢呋喃及其杂质r-对映异构体((r)-(+)-3-羟基四氢呋喃)衍生变为(s)-(+)-3-乙酰氧基四氢呋喃及(r)-(+)-3-乙酰氧基四氢呋喃,然后再采用气相色谱法测定,通过检测(s)-(+)-3-羟基四氢呋喃衍生溶液中的(r)-(+)-3-乙酰氧基四氢呋喃含量,能够准确的测定(s)-(+)-3-羟基四氢呋喃中对映异构体杂质(r)-(+)-3-羟基四氢呋喃含量。
本发明的技术方案是:
在催化剂n,n-二甲基氨基吡啶(dmap)和缚酸剂三乙胺存在下,向(s)-(+)-3-羟基四氢呋喃中添加乙酸酐衍生化(s)-(+)-3-羟基四氢呋喃和其中杂质r-对映体((r)-(+)-3-羟基四氢呋喃)60min,然后用气相色谱法进行测定,能够准确的测定其含量。
衍生化衍生机理如下:
具体的,本发明所述(s)-(+)-3-羟基四氢呋喃中对映异构体杂质(r)-(+)-3-羟基四氢呋喃的检测方法,包括以下步骤:
第一步:样品处理(衍生及检测液配制):
(1)供试品溶液的配制:称取(s)-(+)-3-羟基四氢呋喃90-110mg置5ml量瓶中,加2-3ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶(dmap)5.4-6.6mg、三乙胺207-253mg、乙酸酐0.25-0.3ml,振摇溶解,用乙酸乙酯定容,摇匀,在室温条件下放置1小时,备用。
优选的,称取(s)-(+)-3-羟基四氢呋喃100mg置5ml量瓶中,加2.5ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶(dmap)6.0mg、三乙胺230mg、乙酸酐0.27ml,振摇溶解,用乙酸乙酯定容,摇匀,在室温条件下放置1小时,备用。
(2)对照品溶液的配制:取(r)-(+)-3-羟基四氢呋喃90-110mg置5ml量瓶中,加2-3ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶(dmap)5.4-6.6mg、三乙胺207-253mg、乙酸酐0.25-0.3ml,振摇溶解,用乙酸乙酯定容,摇匀,在室温条件下放置1小时,备用。
第二步:检测:
色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);
升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至120℃,维持2min,再以30℃每分钟的速率升温至150℃,维持5分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;
检测器(fid)温度为250℃;
样品检测:精密量取第一步制备的供试品溶液和对照品溶液各0.5μl,注入气相色谱仪,记录色谱图;面积归一化法计算相应成分的含量。
附图说明
图1为本发明对照例1色谱图;
图2为本发明对照例2色谱图;
图3为本发明实施例1色谱图;
图4为本发明实施例2(乙酸酐定位)色谱图;
图5为本发明实施例2(n,n-二甲基氨基吡啶定位)色谱图;
图6为本发明实施例2(三乙胺定位)色谱图;
图7为本发明实施例2((s)-(+)-3-羟基四氢呋喃定位)色谱图;
图8为本发明实施例2((r)-(+)-3-羟基四氢呋喃定位)色谱图;
图9为本发明实施例2专属性色谱图;
本发明筛选、对照例及实施例用(s)-(+)-3-羟基四氢呋喃样品(来源:安润医药科技(苏州)有限公司,批号:20170302)。
具体检测条件筛选:
衍生时间筛选:
检测条件:色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至120℃,维持2min,再以30℃每分钟的速率升温至150℃,维持5分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;检测器(fid)温度为250℃;进样量为0.2μl。
样品配制:称取(s)-3-羟基四氢呋喃约100mg置5ml量瓶中,加2ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶约6mg、三乙胺约230mg、乙酸酐0.27ml,振摇溶解,用乙酸乙酯定容,摇匀,作为衍生化溶液在室温下放置0min、30min、60min分别进样;检测衍生液中(s)-(+)-3-羟基四氢呋喃含量,测定结果见表1。
表1测定结果
表1结果说明:衍生时间为第60min的衍生溶液中,剩余(s)-(+)-3-羟基四氢呋喃的量为0,即在第60min,完全衍生成(s)-(+)-3-乙酰氧基四氢呋喃。故衍生时间选择60min。
色谱柱筛选:
样品配制:称取(s)-(+)-3-羟基四氢呋喃及其对照品(r)-(+)-3-羟基四氢呋喃各约100mg分别置5ml量瓶中,加2ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶约6mg、三乙胺约230mg、乙酸酐0.27ml,振摇溶解,用乙酸乙酯定容,摇匀,作为供试品溶液及对照品溶液在室温下放置60min进样。
检测条件:色谱柱:气相手性色谱柱rt-bdexm(规格:30m×0.25mm×0.25µm);
升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至120℃,维持2min,再以30℃每分钟的速率升温至180℃,维持6分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;检测器(fid)温度为250℃;进样量为0.2μl,测定(s)-(+)-3-羟基四氢呋喃样品中(r)-(+)-3-乙酰氧基四氢呋喃与(s)-(+)-3-乙酰氧基四氢呋喃的含量,结果见表2。
表2.测定结果
表2结果说明采用rt-bdexm色谱柱供试品与对照品峰重叠,而采用cp-chirasildexcb色谱柱二者分离度为0.79,可基线分离证明采用气相手性色谱柱cp-chirasildexcb优于rt-bdexm。
进样量筛选:
样品配制:称取(s)-(+)-3-羟基四氢呋喃样品约100mg分别置5ml量瓶中,加2ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶约6mg、三乙胺约230mg、乙酸酐0.27ml,振摇溶解,用乙酸乙酯定容,摇匀,作为供试品溶液及对照品溶液在室温下放置60min进样。进样量分别为0.3μl、0.5μl、1.0μl,检测条件:色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);
升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至120℃,维持2min,再以30℃每分钟的速率升温至150℃,维持5分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;检测器(fid)温度为250℃,分别测定(s)-(+)-3-羟基四氢呋喃衍生样品中(r)-(+)-3-乙酰氧基四氢呋喃与(s)-(+)-3-乙酰氧基四氢呋喃的含量,结果见表3。
表3测定结果
表3结果说明:进样量0.5μl时两峰可达基线分离,且分离度优于1.0μl,响应(峰高)优于0.3μl,故进样量优选0.5μl。
有益效果:本发明提供了一种衍生化方法,以测定(s)-(+)-3-羟基四氢呋喃中杂质r-对映异构体含量。本发明技术方案操作方便、衍生时间短、且具有较好的分离情况、精密度,以及很好的重复性。
具体实施例:
实施例1(最优衍生剂及催化剂)
样品配制:称取(s)-(+)-3-羟基四氢呋喃105.17mg,加2ml乙酸乙酯溶解,再称取n,n-二甲氨基吡啶5.88mg、三乙胺229.75mg、乙酸酐0.27ml置同一5ml量瓶中,用乙酸乙酯溶解并稀释至刻度,摇匀,室温放置60min。
检测条件:色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至120℃,维持2min,再以30℃每分钟的速率升温至150℃,维持5分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;检测器(fid)温度为250℃;进样量为0.2μl。
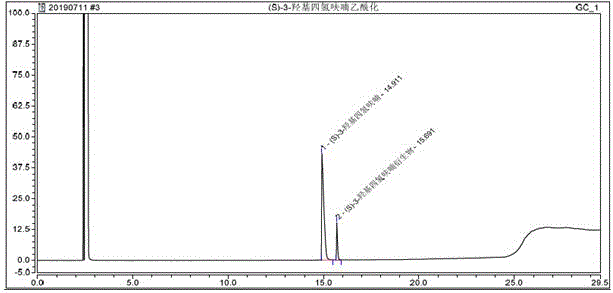
测试结果:衍生剂为乙酸酐,缚酸剂三乙胺和催化剂n,n-二甲基氨基吡啶,(s)-(+)-3-羟基四氢呋喃保留时间处未检出峰,说明(s)-(+)-3-羟基四氢呋喃完全衍生,色谱图见附图3。
实施例2(专属性)
色谱条件:色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);
升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至120℃,维持2min,再以30℃每分钟的速率升温至150℃,维持5分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;检测器(fid)温度为250℃;
样品配制:定位溶液:取(s)-(+)-3-羟基四氢呋喃(100μl)、(r)-(+)-3-羟基四氢呋喃(100μl)、乙酸酐(100μl)、三乙胺(约100mg)、n,n-二甲基氨基吡啶(约6mg)分别置10ml量瓶中加乙酸乙酯溶解稀释至刻度,作为各定位溶液;取(r)-(+)-3-羟基四氢呋喃约100mg置5ml量瓶中,加2ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶约6mg、三乙胺227.81mg、乙酸酐0.27ml,振摇溶解,用乙酸乙酯定容,摇匀,在室温条件下放置1小时后,进样作为(r)-(+)-3-乙酰氧基四氢呋喃定位溶液;称取(s)-(+)-3-羟基四氢呋喃约100mg置5ml量瓶中,加2ml乙酸乙酯溶解,加n,n-二甲基氨基吡啶约6mg、三乙胺约230mg、乙酸酐0.27ml,振摇溶解,用乙酸乙酯定容,摇匀,直接进样作为专属性溶液。
分别进样空白溶剂和所有的定位溶液来确定所有已知成分,进样专属性溶液确定待测杂质色谱峰与相邻的色谱峰之间的分离度不得小于0.5。测定结果见表4。
表4(s)-(+)-3-羟基四氢呋喃手性异构体检测方法专属性考察测定结果
表4数据说明:
实施例3(杂质添加试验考察回收率)
色谱条件:同实施例2色谱条件
样品配制:溶剂:乙酸乙酯
(s)-(+)-3-羟基四氢呋喃贮备液:称取(s)-(+)-3-羟基四氢呋喃504.70mg,置5ml量瓶中,加乙酸乙酯溶解并稀释至刻度,摇匀。
(r)-(+)-3-羟基四氢呋喃贮备液1:称取(r)-(+)-3-羟基四氢呋喃对照品109.49mg,置5ml量瓶中,加乙酸乙酯溶解并稀释至刻度摇匀。
(r)-(+)-3-羟基四氢呋喃贮备液2:精密量取(r)-(+)-3-羟基四氢呋喃贮备液11ml置100ml量瓶中,用乙酸乙酯稀释至刻度,摇匀。
供试品溶液:精密量取(s)-(+)-3-羟基四氢呋喃贮备液1ml,再加入2ml乙酸乙酯,加入n,n-二甲基氨基吡啶6.69mg、三乙胺228.73mg、乙酸酐0.27ml置同一5ml量瓶中,用乙酸乙酯溶解并稀释至刻度,摇匀。
供试品溶液(添加0.1%r异构体):精密量取(s)-(+)-3-羟基四氢呋喃贮备液1ml及(r)-(+)-3-羟基四氢呋喃贮备液21ml,置同一5ml量瓶中,再加入2ml乙酸乙酯,加入n,n-二甲基氨基吡啶6.78mg、三乙胺231.20mg、乙酸酐0.27ml,用乙酸乙酯溶解并稀释至刻度,摇匀。
供试品溶液(添加1%r异构体):精密量取(s)-(+)-3-羟基四氢呋喃贮备液1ml及(r)-(+)-3-羟基四氢呋喃贮备液10.1ml,置同一5ml量瓶中,再加入2ml乙酸乙酯,加入n,n-二甲基氨基吡啶6.01mg、三乙胺233.00mg、乙酸酐0.27ml,用乙酸乙酯溶解并稀释至刻度,摇匀。将上述样品放置60min注入气相色谱仪,按照实施例2色谱条件进行检测。
测定结果见表5。
表5回收率试验测定结果
表5结果说明:将对照品按0.1%、1.0%限度添加进供试品后进行衍生,结果回收率可达97%-100%,证明回收率良好。
实施例4(重复性)
色谱条件:同实施例2色谱条件;
样品配制:精密称取(s)-(+)-3-羟基四氢呋喃约100mg,置5ml量瓶中,加2ml乙酸乙酯溶解,再加n,n-二甲基氨基吡啶约6mg、三乙胺约230mg、乙酸酐0.27ml,加乙酸乙酯定容,摇匀,在室温条件下放置1小时后,直接进样(同法配制6份样品),测定结果见表6。
表6重复性试验测定结果
表6数据说明:实验结果显示,6次测定结果,(r)-(+)-3-乙酰氧基四氢呋喃杂质含量均为0.14%,rsd为0,符合规定,证明该方法具有一定的精密度。
实施例5(不同批次样品检测)
色谱条件:同实施例2色谱条件;
样品配制:精密称取(s)-(+)-3-羟基四氢呋喃(3批与方法开发不同批次样品)约100mg置5ml量瓶中加入3ml乙酸乙酯溶解,再加入n,n-二甲基氨基吡啶约6mg、三乙胺约230mg、乙酸酐0.25ml,用乙酸乙酯定容,摇匀,在室温条件下放置1小时后,直接进样。测定结果见表7。
表7不同批次样品测定结果
表7结果说明:实验结果显示,3批不同批次样品采用本发明检测方法,能有效将样品(s)-(+)-3-羟基四氢呋喃及其中r对映体杂质((r)-(+)-3-羟基四氢呋喃)衍生化生成(s)-(+)-3-乙酰氧基四氢呋喃和(r)-(+)-3-乙酰氧基四氢呋喃,再采用色谱条件检测出(s)-(+)-3-乙酰氧基四氢呋喃中对映异构体杂质((r)-(+)-3-乙酰氧基四氢呋喃)含量,故达到检测(s)-(+)-3-羟基四氢呋喃及其中r对映体杂质((r)-(+)-3-羟基四氢呋喃)的目的。
对照例:我们使用活性较好的乙酰氯作为衍生化试剂,比较衍生化情况。
对照例1(衍生剂采用乙酰氯反应)
样品配制:称取(s)-(+)-3-羟基四氢呋喃样品103.2mg,称取乙酰氯135.1mg置同一10ml量瓶中,用二氯甲烷溶解并稀释至刻度,摇匀,室温放置60min。
检测条件:色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);
升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至150℃,再以40℃每分钟的速率升温至190℃,维持4分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;
检测器(fid)温度为250℃;进样量为0.2μl。
测试结果:(s)-(+)-3-羟基四氢呋喃衍生程度差,产物(s)-(+)-3-乙酰氧基四氢呋喃含量为13.64%,(s)-(+)-3-羟基四氢呋喃86.36%均未衍生,色谱图见附图1。下一步考虑加入缚酸剂三乙胺,结合乙酰氯反应产生的盐酸促进反应进行。
对照例2(衍生剂乙酰氯基础加入缚酸剂三乙胺反应)
样品配制:称取(s)-(+)-3-羟基四氢呋喃样品102.2mg,称取乙酰氯143.6mg,再称取三乙胺230.6mg置同一10ml量瓶中,用二氯甲烷溶解并稀释至刻度,摇匀,室温放置60min。
检测条件:色谱柱:气相手性色谱柱,cp-chirasildexcb(规格:25m×0.25mm×0.25µm);
升温程序:起始温度为60℃,维持2分钟,以每分钟4℃的速率升温至150℃,再以40℃每分钟的速率升温至190℃,维持4分钟;进样口温度为220℃;分流比为50:1;载气流速为1.0ml/min;
检测器(fid)温度为250℃;进样量为0.2μl。
测试结果:加入缚酸剂三乙胺后衍生程度有所改善,但是反应剧烈冒烟且衍生化程度(产物(s)-(+)-3-乙酰氧基四氢呋喃含量)只有80%左右,乙酰氯性质活泼反应剧烈,试验安全性低,色谱图见附图2。考虑乙酸酐比乙酰氯化学性质稳定,故下一步计划采用乙酸酐作为衍生剂,但是由于乙酸酐化学活性低于乙酰氯加入缚酸剂三乙胺后反应程度理论上比乙酰氯加三乙胺还要差,若采用乙酸酐作为衍生剂,加入缚酸剂三乙胺还需加入催化剂n,n-二甲基氨基吡啶促进反应,测试结果反应完全,故选用乙酸酐作为衍生剂,三乙胺作为缚酸剂,n,n-二甲基氨基吡啶作为催化剂,具体试验见实施例1。
- 还没有人留言评论。精彩留言会获得点赞!