一种药物的一测多评检测方法与流程
1.本发明涉及医药分析化学领域,特别涉及一种药物的一测多评检测方法。
背景技术:
2.蒲参胶囊用于高血脂的血瘀证。血瘀证,指指瘀血内阻,以疼痛,肿块,出血,舌紫,脉涩等为主要表现的证候。凡离开经脉的血液,未能及时排出或消散,而停留于某一处;或血液运行受阻,壅积于经脉或器官之内,失却生理功能者,均属瘀血。气血运行受阻,不通则痛故刺痛、固定、拒按;夜间血行缓慢,瘀阻加重故夜间疼痛加重;瘀积不散而凝结体表,故肿块青紫,腹内肿块坚硬不移;瘀血阻塞脉络,使血液不能循经运行,溢出脉外故出血紫暗,或夹有血块;瘀血阻络,血行障碍,全身得不到气血的温煦濡养,故面色黧黑,口唇、舌体、指甲青紫色暗;瘀久不消,营血不能濡养故肌肤甲错;瘀血内阻,冲任不通故经闭;血脉不通,血不循经,则崩漏。瘀血内阻,血行受阻故丝状红缕,腹壁青筋显露,脉细涩,或结、代,或无脉。
3.蒲参胶囊,由何首乌、蒲黄、丹参、川穹、山楂、泽泻、党参提取物组成。蒲参胶囊的主要成分是何首乌。何首乌(polygonum multiflorum thunb.)为我国传统的中草药之一,生首乌系蓼科植物何首乌的干燥块根,具有解毒、消痈、截疟、润肠通便之功效具有活血化瘀,滋阴化浊的功效。活血祛瘀,滋阴化浊。蒲参胶囊用于高血脂症的血瘀证。症见头晕目眩、头部刺痛、胸部刺痛、胸闷憋气、心悸怔忡、肢体麻木;舌质紫暗或有瘀点,脉象细涩患者。
4.中药材的质量好坏是直接影响中药制剂临床疗效的关键原因之一,我国是天然药物品种最多、蕴藏量最大的国家,也是天然药物人工栽培历史最早的国家。目前中药材的来源较为复杂,且品种繁多,就何首乌而言道地产地就有四川,贵州,重庆,广州,广西,浙江,甘肃等地区,所以许多药材市场常常以次充好从中取得大量的经济利益。此外,现今许多药材种植农户没有统一种植标准,相同地区种源的不同,种植方式的不同对最后的药材质量有着至关重要的影响。特别是过量使用化肥,大幅度提升家中药材的产量,从而导致这与野生药材的气味发生了改变,从而影响了中药的疗效。
5.随着我国中药制剂的快速发展,何首乌的入药问题也受到了人们的广泛关注,何首乌分为生何首乌和制何首乌,由于生何首乌对肝脏有一定的毒副总用,其质量的优劣会直接影响到医疗质量和患者生命安危。
6.因此,为了更好地对药品质量进行控制,保证其临床疗效,需建立全面评价该制剂质量的方法。目前关于选自何首乌提取物、蒲黄提取物、丹参提取物、川穹提取物、山楂提取物、泽泻提取物、党参提取物一种或多种混合物或蒲参胶囊的药物及药材的质量控制方法尚无明确记载,因此提供一种药物及药材的质量控制方法具有重要的现实意义。
技术实现要素:
7.本发明要解决的技术问题是提供一种药物的一测多评检测方法,该方法可以用于
检测蒲参胶囊的10种化学成分,并且可以检测选自党参、川芎、赤芍、泽泻、丹参、山楂、蒲黄、何首乌中一种或多种提取物中的蒽醌类成分。
8.一方面,本发明提供的一种药物的一测多评检测方法,采用高效液相色谱法,色谱条件为:
9.色谱柱:c18液相色谱柱;
10.流动相a:含酸水溶液;
11.流动相b:有机溶剂溶液;
12.采用梯度洗脱程序洗脱;所述酸选自磷酸、甲酸、冰乙酸、三氟乙酸中的一种或多种;所述有机溶剂溶液为乙腈、甲醇、乙醇、异丙醇溶液,优选为甲醇溶液;
13.所述药物为选自何首乌提取物、蒲黄提取物、丹参提取物、川穹提取物、山楂提取物、泽泻提取物、党参提取物一种或多种混合物。
14.所述方法的梯度洗脱程序为:
[0015][0016]
或者
[0017][0018]
或者
[0019][0020]
检测波长为250~296nm和/或320~340nm,优选为254~292nm和/或320~340nm,优选为260~286nm和/或325~335nm,进一步优选为273nm和/或330nm。
[0021]
所述检测方法的含酸水溶液中的酸体积百分数为0.05%~0.5%,优选为0.05%~0.2%,进一步优选为0.1%。所述检测方法的流动相流速为0.5~1.5ml/min,优选为1.0ml/min;柱温为10℃~40℃,优选为15℃~35℃,进一步优选为25℃。
[0022]
所述检测方法可以检测10种化学成分,这10种化学成分包括没食子酸,二苯乙烯苷,大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚。
[0023]
所述检测方法包括以下步骤:
[0024]
(1)对照品溶液的制备:称取没食子酸,二苯乙烯苷,大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚中的一种或多种,加第一溶剂制成对照品溶液;
[0025]
(2)供试品溶液的制备:取供试品内容物,与第二溶剂混合,超声混匀,过滤,得到供试品溶液;
[0026]
(3)色谱柱洗脱;
[0027]
(4)谱图分析。
[0028]
其中,所述步骤(1)中对照品溶液中没食子酸的含量优选为2.2~5.12μg/ml,二苯乙烯苷的含量优选为13.86~117.00μg/ml,大黄酚-8-o-β-d-葡萄糖苷的含量优选为4.8~11.2μg/ml,大黄素-8-o-β-d-葡萄糖苷的含量优选为5.5~12.9μg/ml,大黄素甲醚-8-o-β-d-葡萄糖苷的含量优选为4.9~11.5μg/ml,芦荟大黄素的含量优选为4.6~10.7μg/ml,大黄酸的含量优选为4.7~11μg/ml,大黄素的含量优选为4.9~11.5μg/ml,大黄酚的含量优选为4.5~10.6μg/ml,大黄素甲醚的含量优选为4.6~10.8μg/ml;所述第一溶剂为有机溶剂,优选为醇类溶剂,优选为甲醇;所述第二溶剂为醇类水溶液或醇类溶剂,优选为乙醇水溶液;第二溶剂体积比例为50%~90%,进一步为70%;以g/ml计,供试品与第二溶剂的比例为1:(40~60),进一步为1:50;供试品成分提取方法包括超声或加热回流,优选为超声;所述步骤(3)中选所述色谱柱选自c18色谱柱,优选自thermo syientific c18色谱柱、agilentsb-c18色谱柱或hypurity c18色谱柱。
[0029]
另一方面,本发明提供一种蒲参胶囊的一测多评检测方法,尤其是10种化学成分
的检测方法,采用高效液相色谱法,色谱条件为:
[0030]
色谱柱:c18液相色谱柱;
[0031]
流动相a:含酸水溶液;
[0032]
流动相b:有机溶剂溶液;
[0033]
采用梯度洗脱程序洗脱,洗脱程序如下:
[0034][0035]
或者
[0036][0037]
或者
[0038][0039][0040]
所述酸选自磷酸、甲酸、冰乙酸、三氟乙酸中的一种或多种;所述有机溶剂溶液优选为甲醇溶液。
[0041]
本发明蒲参胶囊的一测多评检测方法,其检测波长为250~296nm和/或320~340nm,优选为254~292nm和/或320~340nm,优选为260~286nm和/或325~335nm,进一步优选为273nm和/或330nm;流动相流速为0.5~1.5ml/min,优选为1.0ml/min;柱温为10℃~40℃,优选为15℃~35℃,进一步优选为25℃;流动相a含酸水溶液中的酸体积百分数为
0.05%~0.5%,优选为0.05%~0.2%,进一步优选为0.1%。
[0042]
具体而言,本发明提供一种适用于多种药物的一测多评检测方法,所述药物为选自何首乌提取物、蒲黄提取物、丹参提取物、川穹提取物、山楂提取物、泽泻提取物、党参提取物一种或多种混合物;或蒲参胶囊,包括以下步骤:
[0043]
(1)对照品溶液的制备:称取没食子酸,二苯乙烯苷,大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚中的一种或多种,加第一溶剂制成标准品溶液;
[0044]
(2)供试品溶液的制备:取供试品内容物,与第二溶剂混合,超声混匀,过滤,得到供试品溶液;
[0045]
(3)色谱柱洗脱;
[0046]
(4)谱图分析。
[0047]
所述步骤(1),对照品溶液包括没食子酸,二苯乙烯苷,大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚的一种或多种组分。
[0048]
所述步骤(1),标准品溶液中没食子酸的含量优选为2.2~5.12μg/ml,二苯乙烯苷的含量优选为13.86~117.00μg/ml,大黄酚-8-o-β-d-葡萄糖苷的含量优选为4.8~11.2μg/ml,大黄素-8-o-β-d-葡萄糖苷的含量优选为5.5~12.9μg/ml,大黄素甲醚-8-o-β-d-葡萄糖苷的含量优选为4.9~11.5μg/ml,芦荟大黄素的含量优选为4.6~10.7μg/ml,大黄酸的含量优选为4.7~11μg/ml,大黄素的含量优选为4.9~11.5μg/ml,大黄酚的含量优选为4.5~10.6μg/ml,大黄素甲醚的含量优选为4.6~10.8μg/ml;
[0049]
所述步骤(1),对照品溶液溶剂(第一溶剂)为有机溶剂,优选为醇类溶剂,优选为甲醇。
[0050]
所述步骤(2),作为优选,蒲参胶囊供试品溶液的制备方法为:取蒲参胶囊与甲醇水或乙醇水溶液混合,超声提取,冷却,甲醇水或乙醇水溶液补足减失质量,混匀,过滤,取续滤液过滤膜,得到蒲参胶囊供试品溶液。
[0051]
所述步骤(2),所述供试品溶液溶剂(第二溶剂)醇类水溶液或醇类溶剂,优选为乙醇水溶液;第二溶剂体积比例为50%~90%,进一步为70%;以g/ml计,供试品与第二溶剂的比例为1:(40~60),进一步为1:50。
[0052]
所述步骤(2),超声提取的时间为20~40min,进一步为30min;滤膜的孔径为0.22~0.45μm。
[0053]
所述步骤(3),色谱柱选自c18色谱柱,优选自thermo syientific c18色谱柱、agilentsb-c18色谱柱或hypurity c18色谱柱;c18的柱长为250mm,内径为4.6mm,粒径为5μm,以(4.6
×
250mm,5μm)表示。流动相a为含酸水溶液,流动相b为甲醇溶液;优选流动相a含酸水溶液中的酸为磷酸、甲酸、冰乙酸、三氟乙酸或其他酸溶液中的一种或多种混合物,优选为磷酸;含酸水溶液的体积百分数为0.05%~0.5%,优选为0.05%~0.2%,进一步优选为0.1%。
[0054]
所述步骤(3),流动相流速为0.5~1.5ml/min,进一步为1.0ml/min;柱温为10℃~40℃,优选为15℃~35℃,进一步优选为25℃。
[0055]
所述步骤(3),色谱柱的洗脱程序为梯度洗脱,梯度洗脱程序为:
葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚适量,精密称定,加甲醇制成含没食子酸2.94μg/ml、二苯乙烯苷15.84μg/ml、大黄酚-8-o-β-d-葡萄糖苷6.42μg/ml、大黄素-8-o-β-d-葡萄糖苷7.38μg/ml、大黄素甲醚-8-o-β-d-葡萄糖苷6.6μg/ml、芦荟大黄素6.12μg/ml、大黄酸6.30μg/ml、大黄素6.60μg/ml、大黄酚6.10μg/ml、大黄素甲醚6.18μg/ml的混合对照品溶液,即得。
[0066]
(2)供试品溶液的制备:取供试品内容物,研细,与甲醇水或乙醇水溶液混合,超声提取,冷却,甲醇水或乙醇水溶液补足减失质量,混匀,过滤,取续滤液过滤膜,得到蒲参胶囊供试品溶液。
[0067]
(3)色谱柱洗脱程序:选用c18色谱柱,柱温20℃~40℃,流速0.5~1.5ml/min,进样量5~20μl,流动相为0.05%~0.5%酸溶液(a)-甲醇(b),梯度洗脱。
[0068]
一种梯度洗脱程序为,
[0069][0070][0071]
另一种梯度洗脱程序为,
[0072][0073]
另一种梯度洗脱程序为,
[0074][0075]
选择波长250~296nm检测药物中没食子酸、二苯乙烯苷(2,3,5,4'-四羟基二苯乙烯-2-o-β-d-葡萄糖苷)、大黄酚-8-o-β-d-葡萄糖苷、大黄素-8-o-β-d-葡萄糖苷、大黄素甲醚-8-o-β-d-葡萄糖苷、芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚的各成分并可后续计算含量;选择波长320nm~340nm检测二苯乙烯苷并可后续计算含量。
[0076]
(4)谱图分析:根据需要分别分析各检测波长下的色谱图。根据标准品的保留时间标记色谱峰。根据hplc检测结果,计算各对照品线性,计算以大黄素对大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡糖糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚的相对校正因子及各组分含量。
[0077]
hplc检测的理论板数不低于3000。hplc检测的检测波长为250~296nm时,色谱峰较全面且各色谱峰分离较好,各成分线性合格。作为优选,检测波长为273nm;hplc检测的检测波长为320~340nm时,可定性和定量分析。作为优选,检测波长为330nm。
[0078]
根据hplc检测结果,可获得选自何首乌提取物、蒲黄提取物、丹参提取物、川穹提取物、山楂提取物、泽泻提取物、党参提取物一种或多种混合物或蒲参胶囊的成分组成及其含量。
[0079]
本发明得到分离度、重现性均较好的十个对照品图谱,对其中8种蒽醌建立了以大黄素对大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡糖糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚的相对校正因子。通过对照品比对,确定了10个成分:没食子酸、二苯乙烯苷(2,3,5,4'-四羟基二苯乙烯-2-o-β-d-葡萄糖苷)、大黄素对大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡糖糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚的出峰时间。并对其进行定量分析。
[0080]
采用以上检测方法,具有如下有益效果:
[0081]
通过与对照品比对,确定了10个成分,分别为没食子酸、二苯乙烯苷(2,3,5,4'-四羟基二苯乙烯-2-o-β-d-葡萄糖苷)、大黄酚-8-o-β-d-葡萄糖苷、大黄素-8-o-β-d-葡萄糖苷、大黄素甲醚-8-o-β-d-葡萄糖苷、芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚。结果显示本发明提供的检测方法具有良好的精密度、线性关系、稳定性、重复性,回收率高,耐用性好;
[0082]
本发明中检测方法的分离度、重现性均较好,信息全面,可通过大黄素和校正因子计算其他成分;
[0083]
本发明可以用于检测选自何首乌提取物、蒲黄提取物、丹参提取物、川穹提取物、山楂提取物、泽泻提取物、党参提取物一种或多种混合物或蒲参胶囊的10种成分(包括蒽醌类化合物8个指标)。方法快速、简便、准确,可作为全面评价该制剂质量的有效方法之一。
附图说明
[0084]
图1实施例1色谱条件在260nm检测波长下对照品的色谱图
[0085]
图2实施例1色谱条件在260nm检测波长下供试品的色谱图
[0086]
图3实施例2色谱条件在273nm检测波长下对照品的色谱图
[0087]
图4实施例2色谱条件在330nm检测波长下对照品的色谱图
[0088]
图5实施例2色谱条件在273nm检测波长下供试品的色谱图
[0089]
图6实施例2色谱条件在330nm检测波长下供试品的色谱图
[0090]
图7实施例6色谱条件在273nm检测波长下供试品的色谱图
[0091]
图8实施例6色谱条件在273nm检测波长下供试品的色谱图
[0092]
图9对比实施例1色谱条件在270nm检测波长下供试品的色谱图
具体实施方式
[0093]
以下实施例用于说明本发明,但并不对发明本身构成限制。
[0094]
实施例1药物的一测多评检测方法
[0095]
对照品溶液的制备:取对照品精密称取没食子酸,二苯乙烯苷,大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚适量,精密称定,加甲醇制成含没食子酸2.94μg/ml、二苯乙烯苷15.84μg/ml、大黄酚-8-o-β-d-葡萄糖苷6.42μg/ml、大黄素-8-o-β-d-葡萄糖苷7.38μg/ml、大黄素甲醚-8-o-β-d-葡萄糖苷6.6μg/ml、芦荟大黄素6.12μg/ml、大黄酸6.30μg/ml、大黄素6.60μg/ml、大黄酚6.10μg/ml、大黄素甲醚6.18μg/ml的混合对照品溶液,即得。
[0096]
供试品溶液的制备:取蒲参胶囊内容物,研细,取约1g,精密称定,置具塞锥形瓶中,精密加入70%乙醇50ml,称定质量,静置30min,超声提取30min,放冷,再称定质量,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液过0.45μm滤膜,即得。
[0097]
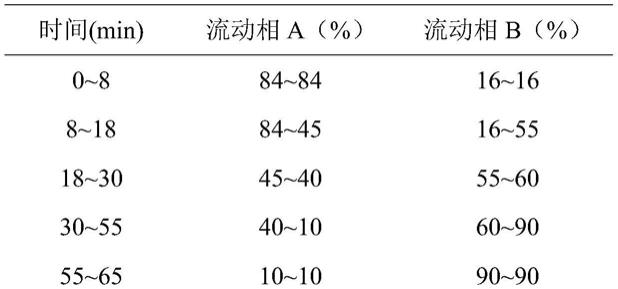
仪器与条件:赛默飞u3000高效液相色谱仪,aglient zoxbar sb-c18色谱柱,检测波长260nm,柱温25℃,流速1.0ml/min,进样量10μl,流动相为0.1%磷酸溶液(a)-甲醇(b),梯度洗脱。
[0098]
洗脱程序见表1。
[0099]
表1实施例1色谱柱梯度洗脱程序
[0100][0101]
对照品和供试品的色谱图分别见图1和图2。图1中1号峰大黄酚-8-o-β-d-葡萄糖苷,2号峰大黄素-8-o-β-d-葡萄糖苷,3号峰为大黄素甲醚-8-o-β-d-葡萄糖苷,4号峰为芦荟大黄素,5号峰为大黄酸,6号峰为大黄素,7号峰为大黄酚,8号峰为大黄素甲醚;图2中1号峰大黄素-8-o-β-d-葡萄糖苷,2号峰为大黄素甲醚-8-o-β-d-葡萄糖苷,3号峰为为大黄素,4号峰为大黄素甲醚。由图可见,蒽醌类对照品和供试品分离度良好。
[0102]
实施例2药物的一测多评检测方法
[0103]
对照品溶液的制备:见实施1对照品溶液的配制。
[0104]
供试品溶液的制备:见实施1供试品溶液的制备。共检测10个批次蒲参胶囊供试品。
[0105]
仪器与条件:赛默飞u3000高效液相色谱仪,aglient zoxbar sb-c18色谱柱,检测波长273nm,柱温25℃,流速1.0ml/min,进样量10μl,流动相为0.1%磷酸溶液(a)-甲醇(b),梯度洗脱。
[0106]
洗脱程序见表2。
[0107]
表2实施例2色谱柱梯度洗脱程序
[0108][0109]
色谱图见图3~6。图3中1号峰是没食子酸,2号峰是二苯乙烯苷,3号峰是大黄酚-8-o-β-d-葡萄糖苷,4号峰是大黄素-8-o-β-d-葡萄糖苷,5号峰是大黄素甲醚-8-o-β-d-葡萄糖苷,6号峰是芦荟大黄素,7号峰是大黄酸,8号峰是大黄素,9号峰是大黄酚,10号峰是大黄素甲醚;图4中1号峰是二苯乙烯苷;图5中1号峰为没食子酸,2号峰为二苯乙烯苷,3号峰为大黄素-8-o-β-d-葡萄糖苷,4号峰为大黄素甲醚-8-o-β-d-葡萄糖苷,5号峰为大黄素,6号峰为大黄素甲醚;图6中1号峰是二苯乙烯苷。
[0110]
其中图5和图6的结果见表3。
[0111]
表3供试品各成分相关液相参数
[0112][0113]
按公式分别计算大黄素对大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚的相对校正因子,结果见表4。
[0114]
表4蒽醌类成分相对校正因子
[0115][0116]
10个批次中蒲参胶囊成分结果见表5。
[0117]
表5蒲参胶囊中各成分含量结果
[0118][0119]
实施例3药材中蒽醌类化合物检测
[0120]
使用本发明实施例2的检测方法,对蒲参胶囊各药材是否含有蒽醌类化合物进行检测,结果见表6。
[0121]
表6蒲参胶囊各药材含蒽醌类化合物情况表
[0122][0123]
实施例4精密度试验
[0124]
采用本发明实施例2的检测方法,取实施例1对照品溶液连续进样6针,以峰面积计算各指标成分的rsd值,分别为没食子酸0.29%、二苯乙烯苷0.09%、大黄酚-8-o-β-d-葡萄糖苷0.43%,大黄素-8-o-β-d-葡萄糖苷0.20%,大黄素甲醚-8-o-β-d-葡萄糖苷0.19%,芦荟大黄素0.56%,大黄酸0.44%,大黄素1.67%,大黄酚1.16%,大黄素甲醚1.33%。由结果可知,该检测方法精密度良好,可操作性强。
[0125]
实施例5线性关系的考察
[0126]
采用本发明实施例2的检测方法,精密称取没食子酸、二苯乙烯苷、大黄酚-8-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,芦荟大黄素,大黄酸,大黄素,大黄酚,大黄素甲醚适量,加甲醇制成含没食子酸5.152μg/ml、二苯乙烯苷27.72μg/ml、大黄酚-8-o-β-d-葡萄糖苷11.235μg/ml,大黄素-8-o-β-d-葡萄糖苷12.915μg/ml,大黄素甲醚-8-o-β-d-葡萄糖苷11.5μg/ml,芦荟大黄素10.71μg/ml,大黄酸11.025μg/ml,大黄素11.55μg/ml,大黄酚10.675μg/ml,大黄素甲醚10.815μg/ml的混合对照品溶液。
[0127]
将其作为母液用甲醇逐倍稀释,分别精密吸取10μl,注入液相色谱仪,采用实施例2提供的检测方法测定,以进样质量浓度为横坐标(x),峰面积为纵坐标(y)进行线性拟合,得线性回归方程见表7。由结果可以看出,该检测方法浓度与峰面积的相关度高,线性关系好,可用来定量分析各成分。
[0128]
表7各成分线性回归方程
[0129]
成分回归方程r没食子酸y=0.3073x+0.00280.9999二苯乙烯苷y=4.2284x+0.28370.9999大黄酚-8-o-β-d-葡萄糖苷y=0.23981x-0.001180.9999大黄素-8-o-β-d-葡萄糖苷y=0.37486x-0.000650.99997大黄素甲醚-8-o-β-d-葡糖糖苷y=0.58222x+0.224350.99993芦荟大黄素y=0.2679x-0.0230.9999大黄酸y=0.3026x-0.01510.9998大黄素y=0.35452x+0.751870.9997大黄酚y=0.24x+0.11090.9999大黄素甲醚y=2.623489x+0.0981300.9999
[0130]
实施例6药物的一测多评检测方法
[0131]
对照品溶液的制备:精密称取大黄素-8-o-β-d-葡萄糖苷12.5mg、大黄素甲醚-8-o-β-d-葡萄糖苷5.8mg于25ml容量瓶,用甲醇定容,作为贮备液1;精密称取大黄素7.2mg于10ml容量瓶,用甲醇定容,做溶液2;精密称取大黄素甲醚15.2mg于25ml容量瓶,用甲醇定容,做溶液3。贮备液:移液管取1ml溶液1,5ml的溶液2,3ml溶液3,用甲醇定容。对照品溶液1:用移液管取10ml贮备液于25ml容量瓶,用甲醇定容即得。对照品溶液2:用移液管取15ml贮备液于25ml容量瓶,用甲醇定容即得。
[0132]
供试品溶液的制备:取蒲参胶囊内容物,研细,取约1g,精密称定,置于25ml容量瓶,加入15ml的70%乙醇,浸泡30min,超声30min,取出放冷至室温,定容,过滤即得。
[0133]
仪器与条件:赛默飞u3000高效液相色谱仪,thermo hypurity-c18色谱柱,检测波长273nm,柱温25℃,流速1.0ml/min,进样量10μl,流动相为0.1%磷酸溶液(a)-甲醇(b),梯度洗脱。
[0134]
洗脱程序见表8。
[0135]
表8实施例6色谱柱梯度洗脱程序
[0136][0137]
色谱图见图7~8。图7和图8中1号峰是二苯乙烯苷,2号峰是大黄素糖苷,3号峰是大黄素甲醚糖苷,4号峰是大黄素,5号峰是大黄素甲醚。
[0138]
其中图8的结果见表9。
[0139]
表9供试品各成分相关液相参数
[0140][0141]
计算公式
[0142][0143]
其中:a
样
:供试品的峰面积
[0144]a对
:对照品的峰面积
[0145]c对
:对照品的浓度
[0146]c样
:供试品的浓度
[0147]w平
:胶囊的平均装量
[0148]
分别计算二苯乙烯苷、大黄素糖苷、大黄素甲醚糖苷、大黄素、大黄素甲醚的含量。结果见表10。
[0149]
表10蒲参胶囊中各成分含量结果
[0150][0151]
实施例7精密度实验
[0152]
采用本发明实施例6的检测方法,精密称取2,3,5,4
’-
四羟基二苯乙烯-2-o-β-d-葡萄糖苷对照品,大黄素-8-o-β-d-葡萄糖苷对照品,大黄素甲醚-8-o-β-d-葡萄糖苷对照品,大黄素对照品,大黄素甲醚对照品,加甲醇溶解制成每1ml分别含有为0.182mg、0.005mg、0.00232mg、0.0365mg、0.01824mg的混合对照品溶液,连续进样6针,记录色谱图,计算rsd%值,结果见表11。
[0153]
表11精密度试验计算结果
[0154][0155]
结论:本品的精密度良好,符合要求。
[0156]
实施例8线性关系考察
[0157]
采用本发明实施例6的检测方法,精密称取2,3,5,4
’-
四羟基二苯乙烯-2-o-β-d-葡萄糖苷对照品,大黄素-8-o-β-d-葡萄糖苷对照品,大黄素甲醚-8-o-β-d-葡萄糖苷对照品,大黄素对照品,大黄素甲醚对照品,加甲醇溶解制成每1ml分别含有为0.364mg、0.01mg、0.00464mg、0.073mg、0.03648mg的混合对照品溶液,进行稀释,见表12,记录色谱图。
[0158]
表12各成分线性回归方程
[0159]
成分回归方程r2二苯乙烯苷y=121.83x-0.00491.0000大黄素糖苷y=372.86x-0.03381.0000大黄素甲醚糖苷y=397.1x-0.00970.9999大黄素y=436.42x-0.01481.0000大黄素甲醚y=354.37x-0.15510.9996
[0160]
实施例9提取方式的考察
[0161]
9.1提取方式的考察
[0162]
精密称取蒲参胶囊内容物1g于25ml容量瓶,选取70%乙醇为溶剂,倒入15ml的70%乙醇,浸泡30min,分别采取超声30min和加热回流30min,进行提取方式的考察。结果见表13。
[0163]
表13不同提取方法下各成分含量结果
[0164][0165]
根据试验结果显示,超声提取与回流提取的含量相差不大,考虑操作便捷性,选择超声作为提取方式。
[0166]
9.2提取溶剂的考察
[0167]
精密称取蒲参胶囊内容物1g于25ml容量瓶,分别选取甲醇和乙醇作为溶剂,分别加入15ml溶剂,浸泡30min,超声30min后取出放凉定容,进行提取溶剂的考察,结果见表14。
[0168]
表14不同提取溶剂下各成分含量结果
[0169][0170]
根据试验结果显示,甲醇提取含量优于乙醇,但是考虑到甲醇乙醇的差异不显著,考虑到环保以及甲醇进样的末端吸收,拟选择乙醇作为提取溶剂。
[0171]
对比实施例1药物的一测多评检测方法
[0172]
对照品溶液的制备:见实施1对照品溶液的配制。
[0173]
供试品溶液的制备:见实施1供试品溶液的制备。
[0174]
仪器与条件:赛默飞u3000高效液相色谱仪,aglient zoxbar sb-c18色谱柱,检测波长270nm,柱温25℃,流速1.0ml/min,进样量10μl,流动相为0.1%磷酸溶液(a)-甲醇(b),梯度洗脱。
[0175]
洗脱程序见表15。
[0176]
表15对比实施例1色谱柱梯度洗脱程序
[0177][0178]
色谱图见图9。供试品中保留时间21min之前的峰不能有效分离。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1