一种用HPLC法检测氨美愈素口服溶液中多种成分含量的方法与流程
本发明涉及药物分析
技术领域:
,具体涉及一种用hplc法检测氨美愈素口服溶液中多种成分含量的方法。
背景技术:
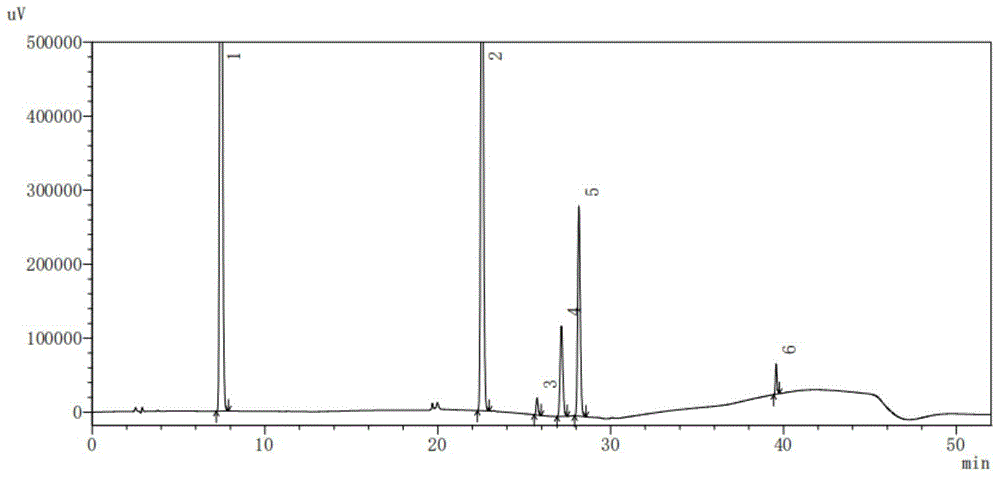
:氨美愈素口服溶液是由活性成分对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素与一定量的非活性成分如助溶剂、矫味剂、防腐剂、抗氧化剂、助氧剂、ph调节剂等辅料组成,是一种治疗儿童多症状感冒发烧药,具有治疗和缓解常见的感冒和流感的症状,如鼻塞、咳嗽和由于轻微咽喉和支气管发炎咳嗽引起冲动的强度,能治疗和缓解轻微疼痛、头痛、喉咙痛,暂时减少发烧,有助于放松痰(黏液)和细支气管分泌物的排出。要保证药品的安全性和有效性,必须进行严格的质量控制。单独的乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素等都有比较成熟的检测方法,例如:对乙酰氨基酚用分光光度法,氢溴酸右美沙芬用高效液相色谱法,愈创木酚甘油醚用滴定法,盐酸去氧肾上腺素用滴定法。但是将其制成复方制剂以后,如果用上述方法分别去检测每一种成份的含量,已经不可行,而且,即使勉强可行,也存在浪费人力物力,浪费资源,污染环境的问题。在现有技术中,也有采用高效液相色谱法同时检测复方制剂中多个成分的方法,但是,很多都具有检测结果不准确的缺点,例如,申请号为cn201410379846.0的利用高效液相色谱分析氨美愈软胶囊的方法,采用含有0.1v%三乙胺的辛烷磺酸钠溶液作为流动相a,以及采用乙腈作为流动相b,用梯度洗脱的方法进行成份分离,虽然也能够一次检测多个成份,但是其采用了三乙胺,是n,n-二乙基乙胺,具有叔胺的化学性质,水溶液呈碱性,与卤代烷反应可生成季铵盐,对氧化剂不稳定。加入辛烷磺酸钠离子对试剂后,又在流动相中加入三乙胺,不但流动相之间要互相影响,也对对乙酰氨基酚、愈创木酚甘油醚产生影响。同时,高效液相的色谱柱多为十八烷基硅烷键合硅胶的填充柱,三乙胺易损伤柱子。申请号为cn201610743206.2的检测药物样品中六个活性成分的方法,所述高效液相色谱法的流动相含有:流动相a,所述流动相a为含有0.1v/v%三氟乙酸的水溶液,以及流动相b,所述流动相b为体积比为60:40的乙腈和甲醇的混合溶液”。虽然也能够一次检测多个成份,但是其具有明显不足。其用到三氟乙酸,而三氟乙酸(tfa)是一种强羧酸,由于含有三氟甲基的特殊结构,其对本发明中的几种主要有效成分都有影响,特别是对对乙酰氨酚影响很大,会使其水解,所以,可能会严重影响检测结果的准确性。技术实现要素:本发明的目的在于提供一种用hplc法检测氨美愈素口服溶液中多种成分含量的方法,解决现有同时测量氨美愈素复方制剂中多种成分方法导致测量结果不准确的问题。本发明通过下述技术方案实现:一种用hplc法检测氨美愈素口服溶液中多种成分含量的方法,所述用hplc法的色谱条件为:以十八烷基硅烷键合硅胶为填充剂,以磷酸盐缓冲溶液和甲醇的混合液为流动相a,以磷酸盐缓冲溶液和乙腈的混合液为流动相b进行梯度洗脱,流速为每分钟0.5-1.5ml,柱温为25-40℃,检测波长为200-254nm。色谱柱的类型、流动相的组成以及流速都会显著影响hplc法有关的保留时间、分离度、拖尾因子等,影响主要成分和杂质检测结果的准确性。申请人通过实验发现:采用上述色谱条件,不仅能够同时检测氨美愈素口服溶液中的对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠和培丙酯,且对其中所有主峰均进行成分指认,具有重现性好,准确率高,适合于本发明氨美愈素口服溶液活性成分含量的质量控制。并且,本发明中所采用的磷酸盐缓冲溶液、甲醇和乙腈均对检测柱、氨美愈素口服溶液中成分没有影响。进一步地,流动相a中磷酸盐缓冲溶液和甲醇的体积比为90:10,所述流动相b中磷酸盐缓冲溶液和乙腈的体积比为25:75。进一步地,梯度洗脱的条件为:0-6分钟,流动相a90-90%、流动相b10-10%;6-20分钟,流动相a90-65%、流动相b10-35%;20-30分钟,流动相a65-50%、流动相b35-50%;30-40分钟,流动相a50-10%、流动相b50-90%;40-55分钟,流动相a90-90%、流动相b10-10%。进一步地,理论板数大于等于1000。进一步地,磷酸盐缓冲溶液为含磷酸二氢钠30mmol/l,辛烷磺酸钠2.50g/l,用磷酸调节ph至3.0的缓冲溶液。进一步地,流速为每分钟1.0ml,柱温为30℃,检测波长为220nm。进一步地,包括以下步骤:s1、对照品溶液的制备:分别称取对照品对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠和培丙酯,然后分别用流动相a溶解并定量稀释制成每1ml中含对乙酰氨基酚325.0μg、氢溴酸右美沙芬10.0μg、愈创木酚甘油醚200.0μg、盐酸去氧肾上腺素5.0μg、苯甲酸钠20.0μg、培丙酯20.0μg的溶液,分别作为对照品溶液;s2、供试品溶液的制备:量取氨美愈素口服溶液,用流动相a定量稀释制成每1ml中含0.01ml氨美愈素口服溶液,过滤,取滤液作为供试品溶液;s3、检测:分别精密吸取对照品溶液5μl和20μl与供试品溶液5μl和20μl,注入液相色谱仪获得色谱图,根据色谱图中的峰面积计算各个成分的含量。口服溶液的具体配方如下:活性成分:对乙酰氨基酚28.0-35.0g、愈创木酚甘油醚15.0-25.0g、盐酸去氧肾上腺素0.4-0.6g、氢溴酸右美沙芬0.8-1.2g;其他辅助组分:1.5-2.5g稳定剂、400.0-600.0g助溶剂、95.0-110.0g矫味剂0.8-1.0g,增稠剂2.5-3.2g,ph调节剂0.4-0.6g,络合剂1.9-2.1g,色素30.0-35.0mg,芳香剂3.0-5.0ml以及防腐剂;加溶剂至总量为1000ml。所述稳定剂为棓丙酯,所述助溶剂为甘油和丙二醇混合物,所述矫味剂为山梨醇和三氯蔗糖的混合物,所述增稠剂为黄原胶,所述ph调解剂为无水枸橼酸和枸橼酸钠的混合物,所述防腐剂为苯甲酸钠,所述络合剂为依地酸二钠,所述芳香剂葡萄香精和蓝莓香精混合物,所述色素为诱惑红色素和亮蓝色素的混合物,所述溶剂为纯化水。本发明与现有技术相比,具有如下的优点和有益效果:1、本发明可以一次检测多个成份,可以实现对乙酰氨基酚、盐酸去氧肾上腺素、愈创木酚甘油醚、氢溴酸右美沙芬以及苯甲酸钠、棓丙酯的同时测定,并且该方法操作简单、分析快捷、重复性好、具有较好的专属性,该分析方法提高了效率,节省了成本。2、避免使用三乙胺,避免了与流动中的烷基磺酸钠产生影响,也避免了对对乙酰氨基酚、愈创木酚甘油醚产生影响。同时,防止了对色谱柱的损伤;避免使用三氟乙酸,解决了其对制剂中几种主要有效成分的影响的问题,使检测结果的准确性大大提高。3、本发明用流动相a作溶剂制备对照品溶液,解决了愈创木酚甘油醚、棓丙酯不溶于水而影响检测结果准确性的问题。4、本发明流动相体系中加入辛烷磺酸钠离子对试剂,增加了盐酸去氧肾上腺素的保留,改善了峰形,增加了分离度;在梯度洗脱的40分钟至40.1分钟让流动相发生较大改变,有效改善了氢溴酸右美沙芬拖尾,并且对不同色谱柱均有很好的耐受性。5、本发明所述的检测方法,能使各组分分离完全,峰形好,供试品测定各组分响应值均在适当的范围,溶剂没有危害。说明书附图图1为对照品溶液色谱图,其中1:对乙酰氨基酚;2:愈创木酚甘油醚;3:盐酸去氧肾上腺素;4:苯甲酸钠;5:棓丙酯;6:氢溴酸右美沙芬。图2为供试品溶液色谱图,其中1:对乙酰氨基酚;2:愈创木酚甘油醚;3:盐酸去氧肾上腺素;4:苯甲酸钠;5:棓丙酯;6:氢溴酸右美沙芬。具体实施方式为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例和附图,对本发明作进一步的详细说明,本发明的示意性实施方式及其说明仅用于解释本发明,并不作为对本发明的限定。实施例:1、色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂(aceexcelc18,4.6mm×250mm,5μm)色谱柱;以磷酸盐缓冲溶液(含磷酸二氢钠30mmol/l,辛烷磺酸钠2.50g/l,用磷酸调节ph至3.0)-甲醇(90:10)为流动相a,以上述磷酸盐缓冲溶液-乙腈(25:75)为流动相b,按表1进行梯度洗脱,流速为每分钟1.0ml,柱温为30℃,检测波长为220nm。表1梯度洗脱2、对照品溶液的制备:取对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、培丙酯适量,精密称定,用流动相a溶解并定量稀释制成每1ml中约含对乙酰氨基酚325.0μg、氢溴酸右美沙芬10.0μg、愈创木酚甘油醚200.0μg、盐酸去氧肾上腺素为5.0μg、苯甲酸钠20.0μg、培丙酯20.0μg的溶液,作为对照品溶液;3、供试品溶液的制备:精密量取氨美愈素口服溶液适量,用上述流动相a定量稀释制成每1ml中约含0.01ml氨美愈素口服溶液,滤过,取续滤液,作为供试品溶液;4、检测:分别精密吸取对照品溶液5μl和20μl与供试品溶液5μl和20μl,注入液相色谱仪,得对照品色谱图(图1)和供试品色谱图(图2),按照外标法以峰面积分别计算,即得氨美愈素口服溶液中的对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠和培丙酯的含量。口服溶液的具体配方如下:活性成分:对乙酰氨基酚28.0-35.0g、愈创木酚甘油醚15.0-25.0g、盐酸去氧肾上腺素0.4-0.6g、氢溴酸右美沙芬0.8-1.2g;其他辅助组分:1.5-2.5g稳定剂、400.0-600.0g助溶剂、95.0-110.0g矫味剂0.8-1.0g,增稠剂2.5-3.2g,ph调节剂0.4-0.6g,络合剂1.9-2.1g,色素30.0-35.0mg,芳香剂3.0-5.0ml以及防腐剂;加溶剂至总量为1000ml。所述稳定剂为棓丙酯,所述助溶剂为甘油和丙二醇混合物,所述矫味剂为山梨醇和三氯蔗糖的混合物,所述增稠剂为黄原胶,所述ph调解剂为无水枸橼酸和枸橼酸钠的混合物,所述防腐剂为苯甲酸钠,所述络合剂为依地酸二钠,所述芳香剂葡萄香精和蓝莓香精混合物,所述色素为诱惑红色素和亮蓝色素的混合物,所述溶剂为纯化水。由图1和图2可以看出:本发明不仅能够同时检测氨美愈素口服溶液中的乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠和培丙酯,且对其中主峰均进行了成分指认,具有准确率高的优点。采用部分方法学研究实验对上述实施方法进行验证:1、系统适用性试验研究:1.1研究样品制备:精密量取对照品适量,用磷酸盐缓冲溶液(磷酸二氢钠30mmol/l,辛烷磺酸钠2.50g/l,用磷酸调节ph至3.0):甲醇=90:10作为流动相a,用流动相a定量稀释制成每1ml中约含对乙酰氨基酚、对氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、棓丙酯分别为325.0μg、10.0μg、200.0μg、5.0μg、20.0μg、20.0μg的溶液,滤过,取20μl注入液相色谱仪,连续进样5次,再取5μl注入液相色谱仪,连续进样5次。1.2研究结果:邻峰最小分离度、理论板数、对称因子、保留时间及峰面积的rsd%如表2所示:表2名称保留时间理论板数分离度对称因子rsd%对乙酰氨基酚7.151145006.7611.1490.03愈创木酚甘油醚2.29312181611.2321.1130.02盐酸去氧肾上腺素25.56629015914.5691.1310.08氢溴酸右美沙芬39.39689830954.2341.1660.11苯甲酸钠26.9421473445.8451.0860.02棓丙酯27.8781863503.4731.1190.02结论:rsd<2,复核要求。2、准确度研究:试验方法:按照120%、100%和80%高中低三个浓度设计试验,分别精密称取对乙酰氨基酚、对氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、棓丙酯,如表3所示,取按处方配置溶液,照上述hplc法测定,浓度样品平行配制三份,照前法试验,计算其回收率,如表4所示:表3表4名称回收率rsd%对乙酰氨基酚100%1.17愈创木酚甘油醚101%1.05盐酸去氧肾上腺素101%1.01氢溴酸右美沙芬98%1.09苯甲酸钠99%0.24棓丙酯99%0.87回收率98-102%、rsd≤2%,复核要求。3、线性研究3.1.样品制备:取对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、棓丙酯对照品,如表5所示,分别置于10ml容量瓶中,加上述流动相a溶解并稀释至刻度,摇匀,制成该系列溶液。分别取20μl注入液相色谱仪,记录色谱图,再分别取5μl注入液相色谱仪,记录色谱图,以峰面积对浓度做标准曲线图。表5研究结果如表6所示:表6r2≥0.999,符合要求。4、精密度-重复性/重现性研究4.1取对乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、棓丙酯对照品适量,精密称定,用流动相a溶解并定量稀释制成每1ml中分别约含325.0μg、10.0μg、200.0μg、5.0μg、20.0μg、20.0μg的溶液,滤过;取氨美愈素口服溶液,平行制备6份供试品,计算样品含量(%),考察该含量测定方法的精密度。4.2研究结果:样品中待测定成分含量和精密度可接受范围参考表7和表8,表7和表8分别为样品中待测定成分含量和精密度rsd可接受范围。表7待测定成分含量重复性(rsd%)重现性100%1210%1.531%240.1%360.01%4810μg/g(ppm)6111μg/g81610μg/kg(ppb1532表8名称rsd%对乙酰氨基酚0.75愈创木酚甘油醚0.68盐酸去氧肾上腺素0.85氢溴酸右美沙芬0.80苯甲酸钠0.67棓丙酯0.88结果:对乙酰氨基酚、愈创木酚甘油醚含量在10%-1%之间;盐酸去氧肾上腺素、氢溴酸右美沙芬含量在0.1%-0.01%之间;苯甲酸钠、棓丙酯含量在1%-0.1%之间,复核要求。5、溶液稳定性试验取对乙酰氨基酚、对氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、棓丙酯对照品,照含量测定方法配制对照品溶液,在第0、12、24、48、72、96、120小时后测定含量,考察溶液的稳定性,结果如表9所示:表9结论:rsd复核要求6、专属性试验研究:6.1试验设计与样品配制方法使用高温、强光、强酸、强碱、氧化以加速本品的降解,以考察本色谱条件的专属性。取氨美愈素口服溶液,照下表方法分别进行高温、强光、强酸、强碱、氧化破坏试验,再取各降解条件下a~h供试品溶液各20μl注入液相色谱仪,记录色谱图,分别取5μl注入液相色谱仪,记录色谱图,有关物质检查方法专属性试验供试样品制备方法如表10所示:表106.2结论:pda检测器检测主成分的最大吸收波长及峰纯度均符合要求。主成分均完全分离,互不干扰。7、耐用性试验研究:取乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠、棓丙酯对照品,照含量测定方法配制,照含量测定方法在以下条件进行测定,计算含量。供试品样品:制剂混合破坏:取专属性破坏样品各2ml混匀,其中混有此浓度下质控限已知杂质加入对氯苯乙酰胺0.00001625mg、对氨基酚0.00001625mg、愈创木酚甘油醚杂质a0.0004mg、愈创木酚甘油醚杂质b0.0004mg、愈创木酚甘油醚杂质c0.0004mg、愈创木酚甘油醚杂质d0.0004mg、盐酸去氧肾上腺素杂质a0.000025mg、盐酸去氧肾上腺素杂c0.000025mg、盐酸去氧肾上腺素杂d0.000025mg、盐酸去氧肾上腺素杂e0.000025mg、氢溴酸右美沙芬杂质a0.00004mg、氢溴酸右美沙芬杂质b0.00004mg、氢溴酸右美沙芬杂质c0.00004mg,0.45μm过滤,弃1ml初滤液,即得。空白辅料混合破坏,取专属性破坏样品各2ml混匀,0.45μm过滤,弃1ml初滤液,即得,试验条件和结果如表11所示:表11结论:对乙酰氨基酚0.06%、愈创木酚甘油醚0.11%、盐酸去氧肾上腺素0.34%、氢溴酸右美沙芬0.13%、苯甲酸钠0.32%、棓丙酯1.03%,rsd值<2%,复核设计要求。综上,本发明不仅能够同时检测氨美愈素口服溶液中的乙酰氨基酚、氢溴酸右美沙芬、愈创木酚甘油醚、盐酸去氧肾上腺素、苯甲酸钠和培丙酯,且对其中主峰均进行成分指认,具有重现性好,准确率高的优点,适合于本发明氨美愈素口服溶液活性成分含量的质量控制。以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1