一种快速检测水性光油中异噻唑啉酮类化合物含量的方法与流程
[0001]
本发明属于理化检验技术领域,涉及一种快速检测水性光油中异噻唑啉酮类化合物含量的方法。
背景技术:
[0002]
水性光油具有毒性小、挥发性低、材料来源光等特点,越来越受到印刷行业的重视。由于水性光油以水为溶剂,易滋生细菌,因此,在水性光油产品的储存和使用过程中会添加杀菌防腐剂。异噻唑啉酮类化合物是一种新型、防腐效果好、毒性低、药效长的杀菌防腐剂,目前许多的光油产品都会添加此类防腐剂。尽管异噻唑啉酮类物质毒性较低,但它与皮肤接触可能引起过敏或皮炎等反应,过量接触还可导致皮肤灼伤。目前,国内外对食品接触材料、玩具、化妆品等产品中相关异噻唑啉酮类杀菌剂的使用均有严格的限制,而光油产品又是印刷在包装材料的最外层、直接和人体接触的首层材料,因此对于光油产品中异噻唑啉酮类化合物的分析具有较高的应用价值和实际意义。
[0003]
水性光油性质较为特殊,常规的用于纸张、玩具、纺织品中异噻唑啉酮的检测方法不具有适用性。这主要是由于以下几点:(1)水性光油中存在大量的高分子树脂,其粘度较大、沸点较高,使用液相色谱仪、液相色谱质谱仪时不能直接进样,系统、色谱柱容易出现堵塞现象,并且,水性光油样品即使通过高速离心后待测溶液依然混浊,样品前处理是一项挑战。(2)由于主要溶剂是水,气相色谱仪、气相色谱质谱仪也难以适用。目前尚未有针对水性光油中异噻唑啉酮类杀菌剂的分析方法,为准确把握水性光油中异噻唑啉酮类物质含量,并更好地控制其安全性,建立水性光油中异噻唑啉酮类物质的测定方法意义重大。
技术实现要素:
[0004]
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,该方法不仅操作便捷、检测时间短、准确性好,更主要是前处理方法非常绿色环保。
[0005]
为实现上述目的及其他相关目的,本发明提供一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,包括:将水性光油样品加入第一试剂超声萃取后获得的萃取液,再加入第二试剂涡旋振荡后离心,取上清液过滤所得的样品溶液,采用超高效液相色谱法(uplc)进行测定,确定样品溶液中异噻唑啉酮类化合物的含量。
[0006]
较佳地,所述异噻唑啉酮类化合物选自2-甲基-4-异噻唑啉-3-酮(mit)、5-氯-2-甲基-4-异噻唑啉-3-酮(cmi)、1,2-苯并异噻唑啉-3-酮(bit)中的一种或多种。优选地,所述异噻唑啉酮类化合物包括2-甲基-4-异噻唑啉-3-酮(mit)、5-氯-2-甲基-4-异噻唑啉-3-酮(cmi)、1,2-苯并异噻唑啉-3-酮(bit)。
[0007]
上述2-甲基-4-异噻唑啉-3-酮(mit)的cas号为2682-20-4,5-氯-2-甲基-4-异噻唑啉-3-酮(cmi)的cas号为26172-55-4,1,2-苯并异噻唑啉-3-酮(bit)的cas号为2634-33-5。
[0008]
较佳地,所述第一试剂选自水、甲醇、乙腈、正己烷、丙酮、乙酸乙酯、叔丁基甲醚中的一种。
[0009]
优选地,所述第一试剂为水。上述水为超纯水。
[0010]
较佳地,所述水性光油样品加入的质量与第一试剂加入的体积之比为0.1-0.5:10-40,g/ml。优选地,所述水性光油样品加入的质量与第一试剂加入的体积之比为0.2:20,g/ml。
[0011]
较佳地,所述超声萃取的时间为20-50min。优选地,所述超声萃取的时间为30min。
[0012]
较佳地,所述第二试剂选自无水mg2so4、n-丙基乙二胺(psa)、活性炭、石墨烯、碳纳米管中的一种或多种。
[0013]
优选地,所述活性炭为碳载量为c18的活性炭。
[0014]
优选地,所述第二试剂为无水mg2so4和n-丙基乙二胺(psa),所述无水mg2so4与n-丙基乙二胺(psa)加入的质量之比为140-160:25。
[0015]
更优选地,所述无水mg2so4与n-丙基乙二胺(psa)加入的质量之比为150:25。
[0016]
较佳地,所述萃取液加入的体积与第二试剂加入的质量之比为2:150-200,ml/mg。优选地,所述萃取液加入的体积与第二试剂加入的质量之比为2:175,ml/mg。
[0017]
较佳地,所述涡旋振荡的时间为2-10min,所述涡旋振荡的频率为500-3000r/min。优选地,所述涡旋振荡的时间为10min,所述涡旋振荡的频率为2000r/min。
[0018]
较佳地,所述离心的时间为2-10min,所述离心的速率为6000-12000r/min。优选地,所述离心的时间为5min,所述离心的速率为11000r/min。
[0019]
较佳地,所述过滤为滤膜过滤,所述滤膜为0.22-0.45μm有机相滤膜。优选地,所述滤膜为0.22μm有机相滤膜。
[0020]
较佳地,所述采用超高效液相色谱仪(uplc)进行测定,包括以下步骤:
[0021]
1)配制标准溶液:取mit、cmi、bit的标准品,加入第一试剂溶解后定容,配成标准溶液;
[0022]
2)样品检测:采用超高效液相色谱法(uplc)分别检测标准溶液和样品溶液,将获得的样品溶液的液相色谱图,与标准溶液的液相色谱图进行比较,根据相对保留时间指认出共有特征峰定性,再根据共有特征峰的色谱峰面积通过外标法定量,从而确定样品溶液中mit、cmi、bit的含量。
[0023]
优选地,步骤1)中,所述标准溶液是将mit、cmi、bit标准品的混合储备溶液,加入第一试剂进行逐级稀释后获得。
[0024]
更优选地,所述混合储备溶液中mit、cmi、bit的浓度均为100μg/ml。
[0025]
优选地,步骤1)中,所述标准溶液中mit、cmi、bit的浓度范围均为0.02-5.0μg/ml。
[0026]
优选地,步骤2)中,所述超高效液相色谱法采用色谱柱为c18色谱柱。更优选地,所述色谱柱为eclipseplus c18 rrhd色谱柱(50mm
×
3.0mm,1.8μm)。
[0027]
优选地,步骤2)中,所述超高效液相色谱法的色谱条件为:检测器为二极管阵列检测器(dad);检测波长:275nm-318nm;流动相为0.05-0.15%甲酸-水-甲醇溶液,其中,a相为0.05-0.15%甲酸-水,b相为甲醇,分析时间为7min,梯度洗脱。
[0028]
更优选地,所述超高效液相色谱法的色谱条件为:检测器为二极管阵列检测器(dad);检测波长:0-2.5min为275nm,2.5-7.0min为318nm;流动相为0.1%甲酸-水-甲醇溶
液,其中,a相为0.1%甲酸-水,b相为甲醇,分析时间为7min,梯度洗脱。
[0029]
所述0.05-0.15%甲酸-水为体积百分比为0.05-0.15%的甲酸水溶液。所述0.1%甲酸-水为体积百分比为0.1%的甲酸水溶液。
[0030]
更优选地,所述梯度洗脱的具体程序为:
[0031]
0-2.5min,a相:b相体积比为75:25-75:25;
[0032]
2.5-2.6min,a相:b相体积比为75:25-50:50;
[0033]
2.6-5.0min,a相:b相体积比为50:50-50:50;
[0034]
5.0-5.1min,a相:b相体积比为50:50-75:25;
[0035]
5.1-7.0min,a相:b相体积比为75:25-75:25。
[0036]
优选地,步骤2)中,所述超高效液相色谱法还包括有以下色谱条件:流速:0.3-0.7ml/min,优选为0.5ml/min;柱温:20-40℃,优选为30℃;进样量:5-15μl,优选为10μl。
[0037]
优选地,步骤2)中,所述外标法包括以下步骤:
[0038]
a)按步骤1)制备一系列不同浓度的标准溶液,分别进行uplc检测,获得mit、cmi、bit的色谱峰面积与对应mit、cmi、bit的浓度之间线性关系,绘制相应的标准工作曲线,计算得到mit、cmi、bit的标准工作曲线的回归方程;
[0039]
b)将样品溶液进行uplc检测,将获得的mit、cmi、bit的色谱峰面积,代入步骤a)中相应的mit、cmi、bit的标准工作曲线的回归方程,计算得到样品溶液中mit、cmi、bit的浓度。
[0040]
更优选地,所述标准工作曲线中,以mit、cmi、bit的色谱峰面积为纵坐标(y轴),其相应mit、cmi、bit的浓度为横坐标(x轴)。
[0041]
如上所述,本发明提供的一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,具有以下有益效果:
[0042]
(1)本发明提供的一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,首次提出了采用超高效液相色谱法测定水性光油中3种异噻唑啉酮类化合物,方法分析速度快、重复性和回收率好,能够在3min以内完成对3种异噻唑啉酮类化合物的检测,适用于大批量样品的快速筛查、分析。
[0043]
(2)本发明提供的一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,使用的超高效液相色谱法相比普通液相色谱法,其灵敏度高、分析时间短、分析效率高;其相比普通液相色谱质谱法,价格便宜、不需要使用同位素内标(同位素内标价格较高)、操作简单、响应稳定性高、检测成本低、方法推广度高;其相比气相色谱/气相色谱质谱法,不需要进行衍生化、不需要除去水后进行检测、避免使用同位素内标、仪器稳定性高。
[0044]
(3)本发明提供的一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,所用的溶剂可均为超纯水,绿色环保、价格低廉;并且,不使用甲醇进行萃取,避免了甲醇萃取引起的液相色谱溶剂效应;同时由于异噻唑啉酮类物质在水溶液中溶解度很高而其他的有机杂质溶解度小,用超纯水作为萃取剂可以减少复杂样品检测时的干扰现象。
[0045]
(4)本发明提供的一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,采用的前处理方式能得到澄清的样品溶液,避免分析水性光油样品时造成液相系统、色谱柱的堵塞,在进行分析测试后不需要要用大量流动相冲洗色谱柱和系统,即节约成本,又绿色环保。同时,净化剂的使用也可帮助减少复杂样品检测时的干扰现象。
附图说明
[0046]
图1显示为本发明的标准溶液中3种异噻唑啉酮类化合物的超高效液相色谱图,图中,a为mit,b为cmi,c为bit。
[0047]
图2显示为本发明的水性光油样品溶液中3种异噻唑啉酮类化合物的超高效液相色谱图,图中,a为mit,b为cmi,c为bit。
具体实施方式
[0048]
下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。
[0049]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0050]
以下实施例使用的试剂和仪器如下:
[0051]
1、试剂
[0052]
2-甲基-4-异噻唑啉-3-酮(mit)、5-氯-2-甲基-4-异噻唑啉-3-酮(cmi)、1,2-苯并异噻唑啉-3-酮(bit)(纯度≥98%,德国dr公司);乙腈、甲醇、甲酸(色谱纯,上海安普实验科技股份有限公司);超纯水(超纯水仪自制,符合gb/t6682规定的一级水)。
[0053]
2、仪器
[0054]
agilent 1290infinity ii超高效液相色谱仪(美国agilent公司,配有二极管阵列检测器);电子天平(感量:0.1mg,瑞士mettler toledo公司);移液器(德国brand公司);5804r高速离心机(德国eppendorf公司);milli-q超纯水仪(美国millipore公司);2700th超声波清洗器(上海安普实验科技股份有限公司);0.22μm有机相滤膜、一次性注射器(上海安普实验科技股份有限公司);各种规格的容量瓶和具塞锥形瓶(德国witeg公司)。
[0055]
对于水性光油中3种异噻唑啉酮类化合物:2-甲基-4-异噻唑啉-3-酮(mit)、5-氯-2-甲基-4-异噻唑啉-3-酮(cmi)、1,2-苯并异噻唑啉-3-酮(bit)的含量,包括以下的测定过程。
[0056]
1、样品溶液的制备
[0057]
取水性光油样品加入第一试剂超声萃取20-50min后获得的萃取液。其中,第一试剂选自水、甲醇、乙腈、正己烷、丙酮、乙酸乙酯、叔丁基甲醚中的一种。水性光油样品加入的质量与第一试剂加入的体积之比为0.1-0.5:10-40,g/ml。
[0058]
再取一定量的萃取液加入第二试剂以500-3000r/min的频率进行涡旋振荡2-10min后,以6000-12000r/min的速率进行离心2-10min,取上清液进行0.22-0.45μm有机相滤膜过滤,获得样品溶液。其中,所述第二试剂选自无水mg2so4、psa、活性炭、石墨烯、碳纳米管中的一种或多种,优选为无水mg2so4和psa,无水mg2so4与psa加入的质量之比为140-160:25。萃取液加入的体积与第二试剂加入的质量之比为2:150-200,ml/mg。
[0059]
2、标准溶液的制备
[0060]
分别准确称取5mg的mit、cmi、bit的标准品于50ml容量瓶内,加入第一试剂溶解后定容,配成mit、cmi、bit的浓度均为100μg/ml的混合储备溶液。再准确称取混合储备溶液加
入第一试剂进行稀释定容后,配成含有浓度范围均为0.02-5.0μg/ml的mit、cmi、bit的标准溶液。第一试剂选自水、甲醇、乙腈、正己烷、丙酮、乙酸乙酯、叔丁基甲醚中的一种。
[0061]
3、测定
[0062]
采用超高效液相色谱法(uplc)分别检测标准溶液和样品溶液,将获得的样品溶液的液相色谱图,与标准溶液的液相色谱图进行比较,根据相对保留时间指认出共有特征峰定性,再根据共有特征峰的色谱峰面积通过外标法定量,从而确定样品溶液中mit、cmi、bit的含量。
[0063]
具体来说,外标法可以将标准溶液选取一系列不同浓度,分别进行uplc检测,获得mit、cmi、bit的色谱峰面积与对应mit、cmi、bit的浓度之间线性关系,绘制相应的标准工作曲线,计算得到mit、cmi、bit的标准工作曲线的回归方程。再将样品溶液进行uplc检测,将获得的mit、cmi、bit的色谱峰面积,代入步骤a)中相应的mit、cmi、bit的标准工作曲线的回归方程,计算得到样品溶液中mit、cmi、bit的浓度。
[0064]
其中,超高效液相色谱法的色谱条件为:色谱柱为c18色谱柱;流速:0.3-0.7ml/min;柱温:20-40℃;进样量:5-15μl;检测器为二极管阵列检测器(dad);检测波长:275nm-318nm;流动相为0.05-0.15%甲酸-水-甲醇溶液,其中,a相为0.05-0.15%甲酸-水,b相为甲醇,分析时间为7min,梯度洗脱。
[0065]
所述梯度洗脱的具体程序为:
[0066]
0-2.5min,a相:b相体积比为75:25-75:25;
[0067]
2.5-2.6min,a相:b相体积比为75:25-50:50;
[0068]
2.6-5.0min,a相:b相体积比为50:50-50:50;
[0069]
5.0-5.1min,a相:b相体积比为50:50-75:25;
[0070]
5.1-7.0min,a相:b相体积比为75:25-75:25。
[0071]
实施例1
[0072]
1、样品溶液的制备
[0073]
取0.2g水性光油样品加入20ml超纯水于三角瓶中,超声萃取30min后获得的萃取液。
[0074]
再取2ml萃取液加入150mg无水mg2so4+25mg psa以2000r/min的频率进行涡旋振荡10min后,以11000r/min的速率进行离心5min,取上清液进行0.22μm有机相滤膜过滤,获得样品溶液1#。
[0075]
2、标准溶液的制备
[0076]
分别准确称取5mg的mit、cmi、bit的标准品于50ml容量瓶内,加入超纯水溶解后定容,配成mit、cmi、bit的浓度均为100μg/ml的混合储备溶液。再分别准确称取20、50、100、200、1000、3000、5000μl的混合储备溶液加入超纯水进行稀释定容至100ml容量瓶后,配成含有浓度分别均为0.02、0.05、0.1、0.2、1.0、3.0、5.0μg/ml的mit、cmi、bit的一系列标准溶液。
[0077]
3、测定
[0078]
采用超高效液相色谱法(uplc)分别检测标准溶液和样品溶液,将获得的样品溶液的液相色谱图,与标准溶液的液相色谱图进行比较,根据相对保留时间指认出共有特征峰定性,再根据共有特征峰的色谱峰面积通过外标法定量,从而确定样品溶液中mit、cmi、bit
的含量,具体见图1、2。
[0079]
具体来说,外标法可以将标准溶液选取一系列不同浓度,分别进行uplc检测,获得mit、cmi、bit的色谱峰面积与对应mit、cmi、bit的浓度之间线性关系,绘制相应的标准工作曲线,计算得到mit、cmi、bit的标准工作曲线的回归方程。再将样品溶液进行uplc检测,将获得的mit、cmi、bit的色谱峰面积,代入相应的mit、cmi、bit的标准工作曲线的回归方程,计算得到样品溶液中mit、cmi、bit的浓度。
[0080]
其中,超高效液相色谱法的色谱条件为:色谱柱为eclipseplus c18 rrhd色谱柱(50mm
×
3.0mm,1.8μm);流速:0.5ml/min;柱温:30℃;进样量:10μl;检测器为二极管阵列检测器(dad);检测波长:0-2.5min为275nm,2.5-7.0min为318nm;流动相为0.1%甲酸-水-甲醇溶液,其中,a相为0.1%甲酸-水,b相为甲醇,分析时间为7min,梯度洗脱。
[0081]
所述梯度洗脱的具体程序为:
[0082]
0-2.5min,a相:b相体积比为75:25-75:25;
[0083]
2.5-2.6min,a相:b相体积比为75:25-50:50;
[0084]
2.6-5.0min,a相:b相体积比为50:50-50:50;
[0085]
5.0-5.1min,a相:b相体积比为50:50-75:25;
[0086]
5.1-7.0min,a相:b相体积比为75:25-75:25。
[0087]
实施例2
[0088]
分别将实施例1的步骤2中配制的一系列不同浓度的标准溶液,以mit、cmi、bit的色谱峰面积为纵坐标(y轴),其相应mit、cmi、bit的浓度为横坐标(x轴),进行uplc分析,得到mit、cmi、bit的回归方程,mit、cmi、bit在0.25~5.0μg/ml范围内呈良好的线性关系,相关系数r2均为0.9999,如表1所示。再利用低浓度的加标浓度平行测定6次,所得测定结果标准偏差的3倍为方法检出限(lod),10倍为方法定量限(loq),获得水性光油样品中,mit、cmi、bit的检出限分别为0.01、0.009、0.006mg l-1
,定量限分别为0.03、0.03、0.02mg l-1
,具有较高的灵敏度。
[0089]
表1
[0090]
化合物回归方程相关系数r2检出限mg l-1
定量限mg l-1
mity=99.8229x-0.00380.99990.0100.03cmiy=68.8019x-0.30150.99990.0090.03bity=156.9870x+0.102450.99990.0060.02
[0091]
注:y:峰面积;x:浓度
[0092]
实施例3
[0093]
精密度是以相对标准偏差的形式表示。平行试验的精密度可用以验证方法的重复性。选取三个加标浓度分别进行6次平行实验,所得结果见表2,在三个加标水平上,结果的相对标准偏差均小于4%,结果表明方法的精密度良好。
[0094]
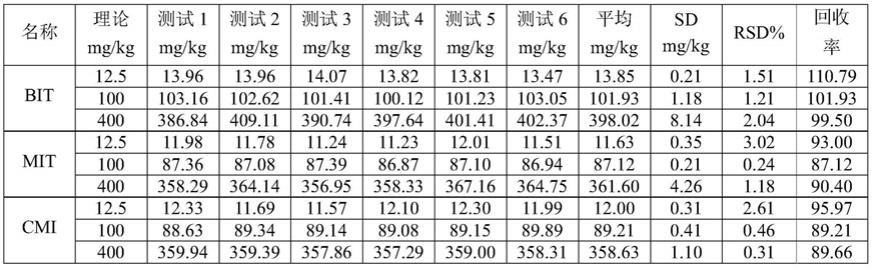
准确度一般通过回收率的结果体现,实验测试了三个加标水平上的6个平行实验的加标回收率的平均值,所得结果见表2。水性光油样品中,低、中、高浓度加标回收率在87.12~110.79%之间,相对标准偏差不高于4%。结果表明,该分析方法对异噻唑啉酮类化合物的测定结果具有准确性。
[0095]
表2油墨样品中准确度和精密度测试结果
[0096][0097]
综上所述,本发明提供的一种快速检测水性光油中异噻唑啉酮类化合物含量的方法,方法分析速度快、重复性和回收率好,减少复杂样品检测时的干扰现象,节约成本,又绿色环保。所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
[0098]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1