对样本免疫组库测序数据进行处理的方法和系统与流程
本发明涉及免疫组库技术领域,具体地,涉及对样本免疫组库测序数据进行处理的方法和系统。
背景技术:
免疫组库(immune reperoire)是指构成机体免疫系统的B细胞受体/抗体(BCR)或T细胞受体(TCR)的集合。B细胞受体/抗体由两条重链和两条轻链构成,T细胞受体由α链β链或者γ链δ链(很少,~5%)组成,在淋巴细胞成熟的过程中,每条链又由多类基因(重链、β链和δ链由V、D、J、C基因重排,轻链、α链和γ链由V、J、C基因重排)重排产生,其中每类基因都有很多个,这样每条链不同类型基因组合达几千到上万种。另外,不同类型基因在重排时,基因之间连接的区域会有不同程度的碱基删除和插入(V,D,J连接端会删除碱基,V-D、D-J或者V-J之间有碱基插入),且删除和插入的碱基随机性很大,这样导致基因重排后,序列的多态性达到惊人的程度。有文章预测人类的α链β链T细胞受体,估计达到了1018,而B细胞受体或抗体的数量会更大,因为B细胞会在此基础上发生单碱基的突变(somatic hyper-mutation)。高通量测序的出现,为研究免疫组库提供了机会。但鉴于如此复杂的基因结构与大数据量,准确的信息分析方法也是一个挑战。
因而,目前的免疫组库研究方法仍有待改进。
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明的一个目的在于提出一种能够有效用于免疫组库研究的手段。
需要说明的是,本发明是基于发明人的下列发现而完成的:
针对免疫组库的分析,目前已有几个工具,如HighV-QUST、IgBLAST、Decombinator,但都存在一些各自的缺陷。HighV-QUST只是在线的工具,分析的数据量有限,不能超过15000条序列,这对高通量的测序数量来说是一个限制。IgBLAST是基因BLAST比对的,但仅仅是做了一个与VDJ基因的比对,没有进一步的处理和统计,Decombinator只能分析TCR数据,不能分析BCR的,并且比对的准确性不是很好。这些工具都没有可视化的图形展示,比如V、J基因的使用频率,CDR3的长度分布,插入删除情况,还有能看到免疫组库多态性的V-J配对图形,缺少这些可视化的图片,影响对免疫组库整体直观反映。发明人认为,免疫组库最重要的分析之一是看序列的频率,一个序列就是一个克隆,如果在序列上发生一个PCR或者测序错率,则会产生一个新的错误的克隆,所以碱基的错误对免疫组库 分析影响很大。但目前已有的这些工具,对PCR和测序错误没有做任何处理,这会很大程度影响结果的准确性。
为了解决上述的问题,本发明在对免疫组库数据进行信息分析时,在基本比对后,会对序列进行重新比对以确定准确的比对结束位置和选出最优比对;对高通量的测序数据,进行PCR和测序错误过滤和纠正;在V(D)J确定后,进行多个数据统计分析,然后以图表的形式展示出来。
根据本发明的一个方面,本发明提供了一种对样本免疫组库测序数据进行处理的方法。根据本发明的实施例,该方法包括:
(1)对样本的免疫组库测序数据进行数据过滤处理,以便获得经过数据过滤处理的测序数据;
(2)将经过数据过滤处理的测序数据进行序列拼接处理,以便获得经过序列拼接处理的测序数据;
(3)将经过序列拼接处理的测序数据分别与V、D、J基因的参考序列进行局部比对,以便获得局部比对结果;
(4)将所述局部比对结果进行重比对,其中将非CDR3编码序列进行全局比对,将CDR3编码序列再次进行局部比对,以便获得重比对结果;
(5)从所述重比对结果中筛选出得分最高的结果,并基于一致性和比对长度对所述得分最高的结果进行过滤,以便获得最终比对结果;
(6)根据测序质量值、序列丰度和V/J基因信息,并基于各读段之间的相似性比较结果,将所述最终比对结果进行纠错处理;以及
(7)基于经过纠错处理的最终比对结果,进行序列结构确定和翻译,以便确定样本的免疫组库信息。
发明人惊奇地发现,利用本发明的方法能够有效实现大数据量的免疫组库测序数据分析,能够一次性分析超过15000条序列,且能够同时满足TCR和BCR数据的分析。此外,本发明的方法能够有效处理PCR和测序错误,并且比对、分析的准确性高,可重复性好,能够有效获得V、J基因的使用频率,CDR3的长度分布,插入删除情况,并且能够有效反映免疫组库多态性的V-J配对情况,甚至能够提供相应的各种可视化图片,从而能够直观反映免疫组库整体情况。
根据本发明的另一方面,本发明还提供了一种对样本免疫组库测序数据进行处理的系统。根据本发明的实施例,该系统包括:
数据过滤装置,所述数据过滤装置用于对样本的免疫组库测序数据进行数据过滤处理,以便获得经过数据过滤处理的测序数据;
序列拼接装置,所述序列拼接装置与所述数据过滤装置相连,用于将经过数据过滤处理的测序数据进行序列拼接处理,以便获得经过序列拼接处理的测序数据;
局部比对装置,所述局部比对装置与所述序列拼接装置相连,用于将经过序列拼接处理的测序数据分别与V、D、J基因的参考序列进行局部比对,以便获得局部比对结果;
重比对装置,所述重比对装置与所述局部比对装置相连,用于将所述局部比对结果进行重比对,其中将非CDR3编码序列进行全局比对,将CDR3编码序列再次进行局部比对,以便获得重比对结果;
筛选过滤装置,所述筛选过滤装置与所述重比对装置相连,用于从所述重比对结果中筛选出得分最高的结果,并基于一致性和比对长度对所述得分最高的结果进行过滤,以便获得最终比对结果;
纠错处理装置,所述纠错处理装置与所述筛选过滤装置相连,用于根据测序质量值、序列丰度和V/J基因信息,并基于各读段之间的相似性比较结果,将所述最终比对结果进行纠错处理;以及
序列结构确定和翻译装置,所述序列结构确定和翻译装置用于基于经过纠错处理的最终比对结果,进行序列结构确定和翻译,以便确定样本的免疫组库信息。
发明人惊奇地发现,利用本发明的系统能够有效实现大数据量的免疫组库测序数据分析,能够一次性分析超过15000条序列,且能够同时满足TCR和BCR数据的分析。此外,本发明的系统能够有效处理PCR和测序错误,并且比对、分析的准确性高,可重复性好,能够有效获得V、J基因的使用频率,CDR3的长度分布,插入删除情况,并且能够有效反映免疫组库多态性的V-J配对情况,甚至能够提供相应的各种可视化图片,从而能够直观反映免疫组库整体情况。
需要说明的是,本发明的方法和系统,具有以下优点的至少之一:
1、与现有数据分析工具相比,本发明分析通量非常高,且适用性更广,广泛适应于TRA、TRB、IGH、IGK、IGL的数据分析,并且适于分析的样品种类不仅仅是人的样品,其他物种的也同样适用。
2、由于V(D)J基因在重排时有不定长度的碱基删除和插入,并且不同的基因之间相似度很高,准确的确定V(D)J有一定的难度,更难的是如何准确的找到基因删除加入的碱基。针对这种情况,本发明在基本的比对后,采用了重比对的步骤,从而能够准确的确定V、D、J基因的比对结束位置,进而准确的确定基因删除和插入碱基,提高了基因比对的准确性。
3、PCR和测序错误对免疫组库的影响很大,但目前的数据分析工具没有去解决这个问题,本发明具有纠正PCR和测序错误的步骤,能够大大降低碱基错误率,相对于现有技术具有明显进步。
4、本发明的方法和系统可以用于很多疾病的免疫组库监控,如白血病微小残留的检测,通过本方法得到的V-J配对三维图,辅助临床技术即能容易地看出病人的康复情况;以及,疫苗的评价,通过本发明的方法可以检测受试者注射前后免疫系统的变化。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
图1和图2分别显示了根据本发明一个实施例,样本1的TRB免疫组库基本数据的部分可视化图;
图3显示了根据本发明一个实施例,样本1中TRB的V-J配对三维图;
图4和图5分别显示了根据本发明一个实施例,样本2的IGH免疫组库基本数据的部分可视化图;
图6显示了根据本发明一个实施例,样本2中IGH的V-J配对三维图;
图7显示了根据本发明一个实施例,对样本免疫组库测序数据进行处理的方法的流程示意图;以及
图8显示了根据本发明一个实施例,对样本免疫组库测序数据进行处理的系统的结构示意图。
具体实施方式
下面详细描述本发明的实施例。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
方法
根据本发明的一个方面,本发明提供了一种对样本免疫组库测序数据进行处理的方法。根据本发明的实施例,该方法包括:
(1)对样本的免疫组库测序数据进行数据过滤处理,以便获得经过数据过滤处理的测序数据;
(2)将经过数据过滤处理的测序数据进行序列拼接处理,以便获得经过序列拼接处理的测序数据;
(3)将经过序列拼接处理的测序数据分别与V、D、J基因的参考序列进行局部比对,以便获得局部比对结果;
(4)将所述局部比对结果进行重比对,其中将非CDR3编码序列进行全局比对,将CDR3编码序列再次进行局部比对,以便获得重比对结果;
(5)从所述重比对结果中筛选出得分最高的结果,并基于一致性和比对长度对所述得分最高的结果进行过滤,以便获得最终比对结果;
(6)根据测序质量值、序列丰度和V/J基因信息,并基于各读段之间的相似性比较结果,将所述最终比对结果进行纠错处理;以及
(7)基于经过纠错处理的最终比对结果,进行序列结构确定和翻译,以便确定样本的免疫组库信息。
发明人惊奇地发现,利用本发明的方法能够有效实现大数据量的免疫组库测序数据分析,能够一次性分析超过15000条序列,且能够同时满足TCR和BCR数据的分析。此外,本发明的方法能够有效处理PCR和测序错误,并且比对、分析的准确性高,可重复性好, 能够有效获得V、J基因的使用频率,CDR3的长度分布,插入删除情况,并且能够有效反映免疫组库多态性的V-J配对情况,甚至能够提供相应的各种可视化图片,从而能够直观反映免疫组库整体情况。
根据本发明的实施例,在步骤(1)中,过滤去除测序接头污染和低质量值的序列。根据本发明的一些具体示例,针对测序接头污染,当接头序列在读段的末端50bp时,切掉该接头序列部分,在序列的其他地方时,则过滤掉整个读段;针对低质量值的序列,当序列末端的碱基测序质量值低于Q10时,切掉该碱基,当序列含有不少于10%的低质量值碱基时,则过滤掉整个读段。由此,有利于后续步骤的进行,能够提高结果的准确性。
根据本发明的实施例,在步骤(2)中,针对插入片段大于单个读长长度的序列,所述序列拼接处理包括:按照给定的最小重叠长度进行拼接,计算重叠区匹配率,再将重叠区域长度逐碱基延长,直到设定的最大重叠长度;选出匹配率最高和第二高的的拼接结果,如果最高匹配率大于设定的值,并且最高匹配率/第二高匹配率的值大于设定值,则输出最好的拼接结果。
根据本发明的另一些实施例,在步骤(2)中,针对插入片段小于单个读长长度的序列,所述序列拼接处理包括:按照给定的最大重叠长度进行拼接,计算重叠区匹配率,再将重叠区域长度逐步减小至给定的最小重叠长度;选出匹配率最高和第二高的的拼接结果,如果最高匹配率大于设定的值,并且最高匹配率/第二高匹配率的值大于设定值,则输出最好的拼接结果。
根据本发明的实施例,进行所述局部比对和重比对可采用的方法和工具不受特别限制。根据本发明的一些具体示例,利用选自BLAST、LASTZ和BLAT的至少一种进行所述局部比对和重比对。由此,比对结果准确可靠。
根据本发明的实施例,在步骤(5)中,针对一个序列,所述得分最高的结果包括:一个得分最高的V基因比对结果、一个得分最高的J基因比对结果。根据本发明的一些具体示例,所述得分最高的结果进一步包括一个得分最高的D基因比对结果。
根据本发明的实施例,在步骤(5)中,基于一致性和比对长度对所述得分最高的结果进行过滤,是过滤掉所述得分最高的结果中一致性小于80%,比对长度小于6bp的读段。由此,能够有效提高数据处理和分析的准确性。
根据本发明的实施例,步骤(6)进一步包括:根据测序质量值,将所述最终比对结果中各读段序列区分为可信的序列和不可信的序列,并将不可信序列与可信序列进行比对,当两者序列之间小于5个错配且都是质量值低的碱基时,则将错配的碱基纠正过来;将所述最终比对结果中的低丰度序列与高丰度序列进行比对,当两者序列之间小于3个碱基错配时,则将低丰度序列上的错配纠正过来。由此,能够有效实现纠错处理,数据处理和分析结果准确性高。
根据本发明的实施例,本发明的方法进一步包括:(8)过滤掉没有比对到V基因或者J基因的序列、比对时V基因和J基因的正负链相反的序列,以及找不到CDR3区域的序列。由此,数据处理和分析结果准确。
根据本发明的实施例,本发明的方法进一步包括:从选自核苷酸、多肽、CDR3的频率分布,V、J、V-J配对的使用率,序列插入片段、CDR3长度分布,插入删除碱基长度分布,体细胞突变率,CDR3种类数和香浓指数值的至少一个方面,对每一步得到的数据进行统计分析;以及将统计分析结果进行作图展示。由此,能够有效提供相应的各种可视化图片,从而能够直观反映免疫组库整体情况。
根据本发明的实施例,本发明的方法进一步包括对每一步得到的数据进行保和性分析的步骤。根据本发明的一些具体示例,可以通过下列公式计算数据的保和性:
其中,
为选取数据量的保和性,
Sobs为实际观测的克隆数目,
F1为选取的数据量中丰度为1的克隆数,
F2为选取的数据量中丰度为2的克隆数。
由此,有利于后续步骤的进行,且能够有效提高各步骤处理结果的准确性。
需要说明的是,将统计分析结果进行作图展示的方法和工具不受特别限制,例如可以采用R语言,SVG等进行。
此外,还需要说明的是,在实施本发明的方法过程中,首先要整理好V(D)J的参考序列,从IMGT(http://www.imgt.org/)下载相应基因的Germline序列,将CDR3的起始或者终止位置在序列上标记好。特别地,对多重PCR,在构建参考序列时应考虑引物序列的影响,若引物与模板存在碱基错配、或者引物与其他模板相似,都应将这些因素考虑进来。
此外,根据本发明的实施例,本发明的方法可以看作四部分,即数据初步处理、V(D)J基因的确定、序列结构分析、数据统计和可视化。进而,根据本发明的一些具体示例,参照图7,本发明的对样本免疫组库测序数据进行处理的方法,还可以包括以下步骤:
1.数据初步处理
a)数据过滤:检查序列是否有测序接头污染,若有则切掉末端污染部分或者过滤掉整个序列。序列末端测序低质量值的碱基被切掉,有较多低质量值碱基的序列会被过滤掉。(这个低质量值是一个参数设置,默认的是Q10)
b)拼接read:对Paired-end的测序类型,将两条reads通过中间重叠的部分拼接起来,成为一条序列。根据插入片段长度大小,分两种情况,对插入片段大于单个read长度的reads,将reads按给定的最小重叠长度(如10bp)进行拼接,计算重叠区match率(完全匹配碱基数/重叠区碱基总数),再将重叠区域长度逐碱基延长,直到设定的最大重叠长度。选出最好和次好的拼接结果(match率最高和第二高),如果最大match率大于设定的值(如90%), 并且match比率(最好match率/次好match率)大于设定值(如0.7),则输出最好的拼接结果。对于插入片段小于单个read长度的reads,拼接时从最大重叠长度逐步减小至最小长度,其他步骤和条件与第一种情况类似。
2.V(D)J基因的确定
a)局部比对:过滤完的序列分别于V、D、J的参考序列进行局部比对(如BLAST)。这里就是用现有的局部比对的软件进行比对,如BLAST,LASTZ,BLAT等等,我们这里用的BLAST,但不局限于这个。全局比对是指序列上的碱基都会去比对,然后得到一个对整个序列来说最优的结果;局部比对,关注是整条序列上的局部,比对得到的是与参考序列最相似的局部序列。
b)重比对:比对得到的结果,进行重新比对,对序列同时用全局比对和局部比对方法结合,非CDR3部分用全局比对的方法,CDR3部分用局部比对的方法。重新计算比对的得分、一致性(identity)、错配数、比对长度、比对的起始终止位置等。(这里没有做过滤,就是所有的比对结果,都会重新计算一个得分、一致性等等)
c)最优比对结果:根据比对的得分,选出得分最高的结果,再根据identity、比对长度进行过滤。一条序列上,会选出一个得分最高的V比对结果、一个D比对结果(如果有D基因的话)、一个J比对结果。一般是identity大于80%,长度大于6bp,不过这两参数可以调整改变。
3.序列结构分析
a)PCR和测序错误纠正:根据测序质量值、序列丰度、V/J基因等信息,序列之间进行相互比较,若相似度高则将mismatch纠正过来。第一步,将序列分类,根据测序质量值,将序列分成可信的序列(质量值高)和不可信的序列;第二步,蒋不可信序列与可信序列比对,如果序列相似度很高(如小于5个错配,且都是质量值低的碱基),则将错配的碱基纠正过来(改为何可信序列一致);第三步,对于低丰度序列(如低于高丰度序列的5倍),与高丰度序列进行比对,如果相似度很高(如小于3个碱基错配),则将低丰度序列上的错配纠正过来。
b)确定序列结构和翻译:标记好比对到假基因或者非功能基因的序列,确定CDR3的区域,根据比对位置和参考序列,确定V(D)J基因末端的碱基删除,以及V-D/D-J或者V-J之间的插入碱基。将核苷酸序列翻译成氨基酸序列,标记好开放阅读框不对和存在终止密码子的序列。
c)过滤:没有比对到V或者J基因的序列、比对时V和J的正负链相反的、找不到CDR3的,这样的序列被过滤掉。一条序列会同时比对上一个V基因和一个J基因,如果比对时V和J基因的正负链不一致,则过滤掉。如V基因为正链比对,J基因为负链比对,则被过滤掉。
4.数据统计和可视化
a)各数据统计分析:每一步数据过滤情况,核苷酸、多肽、CDR3的频率分布,V、J、V-J配对的使用率,序列插入片段、CDR3长度分布,插入删除碱基长度分布,体细胞突变 统计等。这些统计结果基本都有图表展示,都是自己写的R程序实现。
b)保和性分析:随机取不同大小的数据量,统计其克隆数目,包括实际观测到的数量和通过算法预测的最大可能数量。这里的预测算法,是用的生态学中Chao1算法如下:
其中,Sobs实际观测的克隆数目;F1选取的数据量中,丰度为1的克隆数;F2,选取的数据量中,丰度为2的克隆这个分析主要是评价,对于一个样本,我们的目前的测序量是否足够,以及去确定以后需要测多少数据量就已经足够。如果随着选取的数据量增加,预测的最大可能克隆数趋于平缓饱和,则我们的测序量已经够了,并且能知道具体测多少数据量就已经足够;如果预测的值一直在上升未有饱和,则测序量还不够。
c)多态性:用香浓指数计算克隆的多态性。
d)可视化:以上各种统计,都以图表形式体现。其中,V-J配对图是一个三维图形,通过这个图形能反映出整体多样性。
系统
根据本发明的另一方面,本发明还提供了一种对样本免疫组库测序数据进行处理的系统。发明人惊奇地发现,利用本发明的系统能够有效实现大数据量的免疫组库测序数据分析,能够一次性分析超过15000条序列,且能够同时满足TCR和BCR数据的分析。此外,本发明的系统能够有效处理PCR和测序错误,并且比对、分析的准确性高,可重复性好,能够有效获得V、J基因的使用频率,CDR3的长度分布,插入删除情况,并且能够有效反映免疫组库多态性的V-J配对情况,甚至能够提供相应的各种可视化图片,从而能够直观反映免疫组库整体情况。
根据本发明的实施例,参照图8,该系统1000包括:数据过滤装置100、序列拼接装置200、局部比对装置300、重比对装置400、筛选过滤装置500、纠错处理装置600,以及序列结构确定和翻译装置700。
下面结合图8,对本发明的系统1000详细描述如下:
根据本发明的实施例,所述数据过滤装置100用于对样本的免疫组库测序数据进行数据过滤处理,以便获得经过数据过滤处理的测序数据。在所述数据过滤装置100中,过滤去除测序接头污染和低质量值的序列。根据本发明的一些具体示例,针对测序接头污染,当接头序列在读段的末端50bp时,切掉该接头序列部分,在序列的其他地方时,则过滤掉整个读段;针对低质量值的序列,当序列末端的碱基测序质量值低于Q10时,切掉该碱基,当序列含有不少于10%的低质量值碱基时,则过滤掉整个读段。由此,有利于后续步骤的进行,能够提高结果的准确性。
根据本发明的实施例,所述序列拼接装置200与所述数据过滤装置100相连,用于将经过数据过滤处理的测序数据进行序列拼接处理,以便获得经过序列拼接处理的测序数据。
根据本发明的实施例,在所述序列拼接装置200中,针对插入片段大于单个读长长度的序列,所述序列拼接处理包括:按照给定的最小重叠长度进行拼接,计算重叠区匹配率,再将重叠区域长度逐碱基延长,直到设定的最大重叠长度;选出匹配率最高和第二高的的拼接结果,如果最高匹配率大于设定的值,并且最高匹配率/第二高匹配率的值大于设定值,则输出最好的拼接结果。
根据本发明的另一些实施例,在所述序列拼接装置200中,针对插入片段小于单个读长长度的序列,所述序列拼接处理包括:按照给定的最大重叠长度进行拼接,计算重叠区匹配率,再将重叠区域长度逐步减小至给定的最小重叠长度;选出匹配率最高和第二高的的拼接结果,如果最高匹配率大于设定的值,并且最高匹配率/第二高匹配率的值大于设定值,则输出最好的拼接结果。
根据本发明的实施例,所述局部比对装置与所述序列拼接装置200相连,用于将经过序列拼接处理的测序数据分别与V、D、J基因的参考序列进行局部比对,以便获得局部比对结果。
根据本发明的实施例,所述重比对装置400与所述局部比对装置相连,用于将所述局部比对结果进行重比对,其中将非CDR3编码序列进行全局比对,将CDR3编码序列再次进行局部比对,以便获得重比对结果。
根据本发明的实施例,进行所述局部比对和重比对可采用的方法和工具不受特别限制。根据本发明的实施例,所述局部比对装置和所述重比对装置400中,利用选自BLAST、LASTZ和BLAT的至少一种进行所述局部比对和重比对。由此,比对结果准确可靠。
根据本发明的实施例,所述筛选过滤装置500与所述重比对装置400相连,用于从所述重比对结果中筛选出得分最高的结果,并基于一致性和比对长度对所述得分最高的结果进行过滤,以便获得最终比对结果。
根据本发明的实施例,在所述筛选过滤装置500中,针对一个序列,所述得分最高的结果包括:一个得分最高的V基因比对结果、一个得分最高的J基因比对结果。根据本发明的一些具体示例,所述得分最高的结果进一步包括一个得分最高的D基因比对结果。
根据本发明的实施例,在所述筛选过滤装置500中,基于一致性和比对长度对所述得分最高的结果进行过滤,是过滤掉所述得分最高的结果中一致性小于80%,比对长度小于6bp的读段。由此,能够有效提高数据处理和分析的准确性。
根据本发明的实施例,所述纠错处理装置600与所述筛选过滤装置500相连,用于根据测序质量值、序列丰度和V/J基因信息,并基于各读段之间的相似性比较结果,将所述最终比对结果进行纠错处理。
根据本发明的实施例,所述纠错处理装置600适于进一步实施以下步骤:根据测序质量值,将所述最终比对结果中各读段序列区分为可信的序列和不可信的序列,并将不可信序列与可信序列进行比对,当两者序列之间小于5个错配且都是质量值低的碱基时,则将错配的碱基纠正过来;将所述最终比对结果中的低丰度序列与高丰度序列进行比对,当两者序列之间小于3个碱基错配时,则将低丰度序列上的错配纠正过来。由此,能够有效实现纠错处理, 数据处理和分析结果准确性高。
根据本发明的实施例,所述序列结构确定和翻译装置700用于基于经过纠错处理的最终比对结果,进行序列结构确定和翻译,以便确定样本的免疫组库信息。
根据本发明的实施例,本发明的系统1000进一步包括次级过滤装置(图中未示出),所述次级过滤装置与所述序列结构确定和翻译装置700相连,用于过滤掉没有比对到V基因或者J基因的序列、比对时V基因和J基因的正负链相反的序列,以及找不到CDR3区域的序列。由此,数据处理和分析结果准确。
根据本发明的实施例,本发明的系统1000进一步包括统计分析和作图装置(图中未示出),所述统计分析和作图装置适于实施以下步骤:从选自核苷酸、多肽、CDR3的频率分布,V、J、V-J配对的使用率,序列插入片段、CDR3长度分布,插入删除碱基长度分布,体细胞突变率,CDR3种类数和香浓指数值的至少一个方面,对每一步得到的数据进行统计分析;以及将统计分析结果进行作图展示。由此,能够有效提供相应的各种可视化图片,从而能够直观反映免疫组库整体情况。
需要说明的是,如前所述,可以用于将统计分析结果进行作图展示的方法和工具不受特别限制,例如可以采用R语言,SVG等进行。
根据本发明的实施例,各装置均适于进一步对得到的数据进行保和性分析。根据本发明的一些具体示例,通过下列公式计算数据的保和性:
其中,
为选取数据量的保和性,
Sobs为实际观测的克隆数目,
F1为选取的数据量中丰度为1的克隆数,
F2为选取的数据量中丰度为2的克隆数。
由此,有利于后续步骤的进行,且能够有效提高各步骤处理结果的准确性。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件(例如参考J.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》,第三版,科学出版社)或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品,例如可以采购自Illumina公司。
实施例1:
对两个健康人,分别取外周血,从而获得两个外周血样本(样本1和样本2),其中, 样本1用于测TRB,样本2用于测IGH。将外周血样本进行多重PCR扩增,进而构建免疫组库,然后用illumina的Paired-end 100(样本1)和PE150(样本2)进行测序(http://www.illumina.com/),获得两个样本的免疫组库的测序数据。具体地:在TRB/IGH的V、J区设计出多对引物,以便能通过多重PCR扩增捕获所有有功能的重排基因;将外周血提取DNA,与上述设计的引物一起,进行多重PCR反应,PCR进行25个循环;将多重PCR产物进行电泳,回收100-200bp(用于测TRB的样本)片段,和100bp-300bp(用于测IGH的样本)片段,之后进行末端修复和末端加“A”,然后连接测序Adapter,以及进行10个循环的PCR反应来建库;对两个样本的免疫组库进行illumina测序,分别获得免疫组库测序数据。
然后,参照图7,利用本发明的方法进行免疫组库测序数据处理和分析,具体步骤如下:
1.数据初步处理
1)数据过滤:检查序列是否有测序接头污染,若在末端50bp有接头序列,则切掉接头序列部分,若在其他地方,则过滤掉整个序列。序列末端测序质量值低于Q10的碱基被切掉,有较多低质量值碱基的序列会被过滤掉。从下表数据看出,主要过滤掉的是低质量值的序列,最后保留的序列占97%(样本1的TRB)和93%(样本2的IGH),说明整体测序质量还不错。
表1、数据过滤统计表
2)拼接read:对Paired-end的测序类型,将两条reads通过中间重叠的部分拼接起来,成为一条序列。根据插入片段长度大小,分两种情况(插入片段大于单个read长度和插入片段小于read长度)进行处理。步骤上面已给出。表中大部分序列还是长度都大于单个read长度,无法拼接的部分只是占到很小的一部分,说明超过拼接范围的序列含量很少。
表2、read拼接统计表
2.V(D)J基因的确定
表3、V(D)J基因比对统计表
从上图可以看出,能同时找到VJ基因的有98.58%(TRB)进而83.78%(IGH)的序列,若没有同时找到VJ基因,则这样的序列被过滤掉。由于D基因比较短,所以TRB的D基因比对率较低。
3.数据统计和可视化
1)基本数据统计
原始的测序数据,由于有测序接头污染,测序错误、非目标区域捕获等序列的污染,需要对原始数据进行过滤,无法同时比对上VJ的序列被过滤掉。同时,若无法找到多重PCR时的引物,序列也会被丢掉。这里,还有专门的方法纠正PCR和测序错误。对后续的结果分析,只用有功能的序列。CDR3的种类数和香浓指数都是评价样本多态性的重要指标,CDR3数值越大,香浓指数越大,则说明样本多态性越好。
表4、基本数据统计
2)可视化
然后,采用以下可视化R语言程序,对上述统计的基本数据可视化:
结果如下所示:
1)TRB的统计可视化
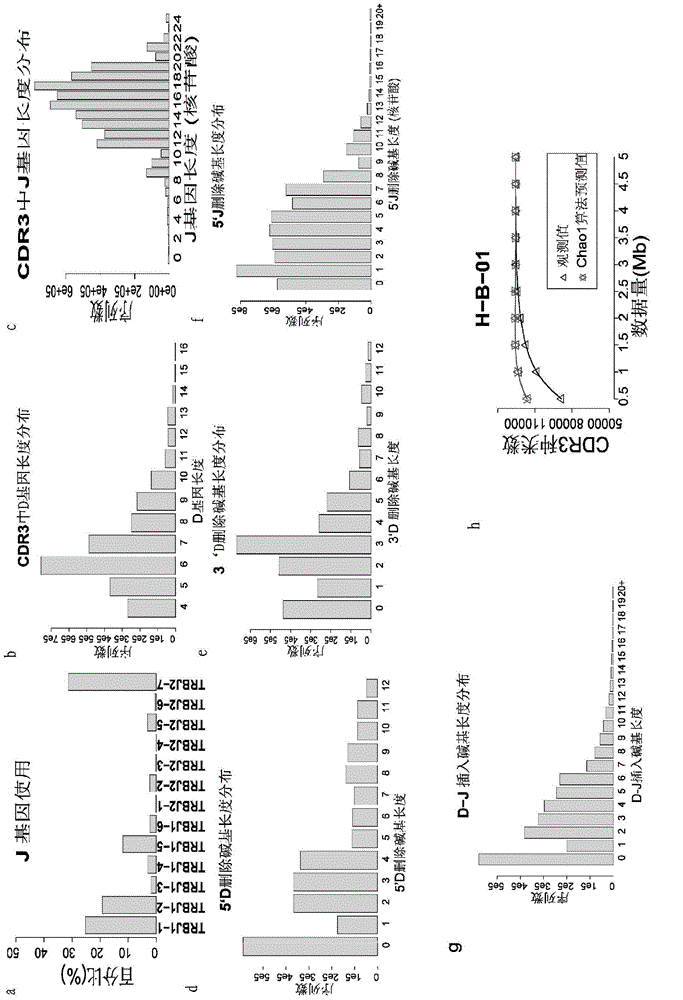
基于表4中样本1的TRB免疫组库基本数据统计结果,进行作图展示,结果见图1-2。其中,图1和图2分别显示了样本1的TRB免疫组库基本数据的部分可视化图。具体地:
如图1所示,各小图分别为:
a.序列长度分布图;b.V基因的使用频率;c.CDR3序列的频率分布图;d.V基因长度分布;e.V基因3‘端的删除长度分布;f.V-D基因之间的插入序列长度分布;g.CDR3长度分布图;h.J基因的碱基组成含量图;i.V-J配对二维分布图。
如图2所示,各小图分别为:
a.J基因的使用频率图;b.D基因的长度分布图;c.J基因的长度分布图;d.D基因5‘的删除碱基分布图;e.D基因的3’的删除碱基分布图;f.J基因的5‘删除碱基分布图;g.D-J之间插入碱基分布图;h.饱和性曲线。
2)TRB的V-J配对三维图
V-J配对三维图是一个直观反映样本多样性的图,如果每个V-J配对的含量都有且叫均匀,则多样性非常好,如果只有几个V-J配对的含量非常高,则反映多样性差,免疫系统的抵抗力可能下降。
发明人基于表4中样本1的TRB免疫组库基本数据统计结果,绘制了样本1中TRB的V-J配对三维图,结果见图3。由图3可知,部分V-J配对含量较低。
3)IGH的统计可视化
基于表4中样本2的IGH免疫组库基本数据统计结果,进行作图展示,结果见图4-5。其中,图4和图5分别显示了样本2的IGH免疫组库基本数据的部分可视化图。具体地:
如图4所示,各小图分别为:
a.序列长度分布图;b.V基因的使用频率;c.CDR3序列的频率分布图;d.V基因长度分布;e.V基因3‘端的删除长度分布;f.V-D基因之间的插入序列长度分布;g.CDR3长度分布图;h.J基因的碱基组成含量图;i.V-J配对二维分布图。
如图5所示,各小图分别为:
a.J基因的使用频率图;b.D基因的长度分布图;c.J基因的长度分布图;d.D基因5‘的删除碱基分布图;e.D基因的3’的删除碱基分布图;f.J基因的5‘删除碱基分布图;g.D-J之间插入碱基分布图;h.饱和性曲线。
4)IGH的V-J配对三维图
发明人基于表4中样本2的TRB免疫组库基本数据统计结果,绘制了样本2中IGH的V-J配对三维图,结果见图6。由图6可知,每个V-J配对都有数据,并且多个V-J配对含量较高,说明多样性比较好。
综合上述结果可知:通过具体实施例(一个TRB和一个IGH)的数据分析,可以明确知道本发明方法每一步的数据分析情况,并且能够清楚的了解本实施例通过数据过滤或统计,在blast比对的基础上,进行重新比对以提高比对的准确性;为减少测序错误和提高有效数据,对PCR和测序错误进行纠正。综合几个体现多态性指标的参数统计,能反映样本的多态性情况。最后,本实施例通过一系列的可视化图形展示,获得了各样本的各种可视化图,基于这些图片能够很直观的了解样本基本数据情况、基因重排的各基因参与情况及插入删除碱基的使用、V-J体现的整体的多样性等等。若受试样本的多样性变得非常少,则很容易从这些图中看出来。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!