一种转录组测序数据计算解读方法与流程
本发明涉及基因测序技术,具体涉及一种转录组测序数据计算解读方法。
背景技术:
近年来,随着下一代测序技术(nextgenerationsequence,ngs)的广泛应用,基因测序的成本迅速下降,基因测序技术得以在更加广泛的生物、医疗、健康、刑侦、农业等等许多领域被推广应用。其中,基于ngs的转录组(transcriptome)测序是一个非常有应用价值的分支领域,受到广泛的关注。
转录组广义上指某一生理条件下,某个物种或者特定细胞类型产生的所有转录本(transcripts)的集合,即各种核糖核酸(ribonucleicacid,rna),包括主要的信使rna(messengerrna,mrna)、核糖体rna(ribosomalrna,rrna)、转运rna(transferrna,trna),还有小分子rna(smallrna,srna),端体酶rna(telomeraserna,terna),反义rna(antisenserna,anrna),以及被称为生命体中“暗物质”的非编码rna(non-codingrna,ncrna),如真核生物中的微rna(microrna,mirna);狭义上指所有mrna的集合。以下描述中,如果未加特殊说明,转录组缺省指代广义转录组。rna是存在于生物细胞以及部分病毒、类病毒中的遗传信息载体,它是以脱氧核糖核酸(deoxyribo-nucleicacid,dna)的一条链为模板,以碱基互补配对原则,转录而形成的一条单链,主要功能是实现遗传信息在蛋白质上的表达,是遗传信息传递过程中的桥梁。rna的碱基主要有4种,即腺嘌呤(adenine,a),鸟嘌呤(guanine,g),胞嘧啶(cytosine,c),和尿嘧啶(uracil,u)。其中,尿嘧啶u取代了dna中的胸腺嘧啶(thymine,t)而成为rna的特征碱基。蛋白质是行使细胞功能的主要承担者,蛋白质组是细胞功能和状态的直接描述,转录组是连接基因组遗传信息与蛋白质组生物功能的必然纽带,转录水平的调控是生物体最重要的调控方式。因此,转录组研究能够从整体水平研究基因功能以及基因结构,揭示特定的生物学过程以及疾病发生过程中的分子机理,对于生物研究、医疗诊断、药物研发等多个领域具有非常大的应用价值。
转录组测序,也称为rna测序(rnasequence,rna-seq)。目前使用ngs技术,能够在单核苷酸(mononucleotide)水平对任意物种及其特定组织或者器官在某一状态下的整体转录活动进行检测,全面快速地获得几乎所有转录本序列信息,为后续的研究打下坚实的基础。相对于传统的芯片杂交平台,基于ngs的转录组测序无需预先针对已知序列设计探针,即可对任意物种及其特定组织或器官在某一状态下的整体转录活动进行检测,而且不存在荧光模拟信号带来的交叉反应和背景噪音问题,能够提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。
基于ngs的转录组测序的数据处理流程包括数据计算和数据解读两大步骤,其中的数据计算步骤完成参考基因组和参考转录组的预处理和原始测序数据的修剪、比对、去重等计算任务,以便数据解读时使用;数据解读步骤对数据计算处理后的数据在生物学、医学、健康保健等领域的科学含义进行分析、揭示和解释。
目前,基于ngs的转录组测序技术在应用上存在两个方面的瓶颈:
一个瓶颈是测序数据产出能力远远大于测序数据处理能力。例如,在基于ngs的转录组测序中比较常用的一个测序数据计算解读协议流程中,参考人类全基因组数据,处理12个样本数据,每个样本数据包含有856k左右、读长为100碱基对(basepair,bp)的双端rna-seq测序片段(reads),使用一台8核、带8gbram、工作主频为2.1ghz的amdopteron6172服务器,进行整个计算解读流程中的一个任务——比对(alignment),就耗时大约12.5小时,而illumina公司的hiseq4000测序仪在5个小时之内能够产出200m个读长为300bp的reads。因此,一方面,测序生成的原始数据每年3到5倍的增加速度已经远超摩尔定律,而测序数据的计算解读又是高输入/输出密集和高计算密集型任务,对测序数据进行实时的、准确的计算解读和传送变得非常困难,面临着巨大的挑战。另一方面,目前典型的测序数据计算解读方法仍然主要是依托高性能的中央处理器(centralprocessingunit,简称cpu),运用基于多线程技术的软件进行处理。但是,在保证准确性的前提下,它能获得的计算解读加速性能仍然无法满足上述挑战的需求。所以,这种方法已经缺乏持续性。
另一个瓶颈是测序数据解读的深度、广度无法满足科研人员的需求,与此同时其可读性又无法满足普通大众的需求。目前测序数据解读的典型方法是基于一个参考基因组,然而,当前使用的参考基因组本身就是基于有限的样本,既不足以代表整个相关物种的多样性,又不完备,因此在数据计算解读时会导致偏差,而且缺乏与其它生物、医学信息的广泛地、深度交叉分析,难以满足专业科研人员深入研究的需求。此外,测序数据解读还基本停留在专业领域,面向非专业的大众,又缺乏可读性,即缺乏对测序数据直接的生物意义和间接的健康影响的通俗易懂、形式多样的解读。
目前,信息处理领域常见的处理器类型有中央处理器(centralprocessingunit,简称cpu)、现场可编程门阵列(fieldprogrammablegatearray,简称fpga)、图形处理器(graphicsprocessingunit,简称gpu)和数字信号处理器(digitalsignalprocessor,简称dsp)。高性能cpu通常都包括多个处理器核(processorcore),从硬件上支持多线程,但是其设计目标还是面向通用应用程序,而相对于特殊的计算,通用应用程序的并行性较小,需要较复杂的控制和较低的性能目标。因此,cpu片上的硬件资源主要还是用于实现复杂的控制而不是计算,没有为特殊功能包含专门的硬件,能够支持的计算并行度不高。fpga是一种半定制电路,优点有:基于fpga进行系统开发,设计周期短、开发费用低;功耗低;生产后可重新修改配置,设计灵活性高,设计风险小。缺点是:实现同样的功能,fpga一般来说比专用集成电路(applicationspecificintegratedcircuit,asic)的速度要慢,比asic电路面积要大。随着技术的发展和演进,fpga向更高密度、更大容量、更低功耗和集成更多硬核知识产权(intellectualproperty,ip)的方向发展,fpga的缺点在缩小,而优点在放大。相比于cpu,fpga可以用硬件描述语言来定制实现、修改和增加并行计算。gpu最初是一种专门用于图像处理的微处理器,能够从硬件上支持纹理映射和多边形着色等图形计算基本任务。由于图形学计算涉及一些通用数学计算,比如矩阵和向量运算,而gpu拥有高度并行化的架构,因此,随着相关软硬件技术的发展,gpu计算技术日益兴起,即gpu不再局限于图形处理,还被开发用于线性代数、信号处理、数值仿真等并行计算,可以提供数十倍乃至于上百倍于cpu的性能。但是目前的gpu存在2个问题:一是,受限于gpu的硬件结构特性,很多并行算法不能在gpu上有效地执行;二是,gpu运行中会产生大量热量,能耗较高。dsp是一种用数字方法对各种信号进行快速分析、变换、滤波、检测、调制、解调等运算处理的微处理器。为此,dsp在芯片内部结构上做了特殊的优化,比如硬件实现高速、高精度的乘法等。随着数字时代的到来,dsp广泛应用于智能设备、资源勘探、数字控制、生物医学、航天航空等各个领域,具有功耗低、精度高、可进行二维与多维处理等特点。综上所述,以上四种计算器件各有特点,又各有局限性。
针对前述基于ngs的转录组测序技术应用发展存在的两个方面的瓶颈,如何利用上述处理器来实现海量测序数据的快速实时、精准深入、通俗易懂、形式多样的计算解读,则已经成为一项亟待解决的关键技术问题。
技术实现要素:
本发明要解决的技术问题:针对现有技术的上述问题,提供一种快速实时、精准深入、通俗易懂、形式多样的转录组测序数据计算解读方法。
为了解决上述技术问题,本发明采用的技术方案为:
一种转录组测序数据计算解读方法,实施步骤包括:
1)输入参考基因组数据、参考转录组数据和原始的测序样本数据,根据预设选择通过cpu调用基于fpga上硬件实现的索引生成器对参考基因组数据、参考转录组数据进行预处理,对原始的测序样本数据进行预处理得到可靠测序样本数据;
2)选择执行基于cpu调用fpga上硬件实现的比对器执行将可靠测序样本数据和参考基因组数据进行比对、将可靠测序样本数据和参考转录组数据进行比对两种任务;
3)基于cpu调用fpga上硬件实现的装配器将可靠测序样本数据重新装配成转录本,选择执行基于cpu调用gpu上编程实现的识别器对可靠测序样本数据和参考基因组数据及其比对结果进行转录本识别、基于cpu调用gpu上编程实现的识别器对可靠测序样本数据和参考转录组数据及其比对结果进行转录本识别;
4)基于cpu调用gpu和dsp将转录组的识别或重新装配的处理结果进行可视化处理,通过cpu调用fpga上硬件实现的深度学习模型在可视化处理得到的结果的基础上进行指定的数据挖掘和分析。
优选地,步骤1)中根据预设选择对参考基因组数据、参考转录组数据进行预处理时,如果需要预处理参考基因组数据,则对参考基因组数据进行预处理的详细步骤包括:读取参考基因组数据,并通过cpu调用基于fpga上硬件实现的索引生成器为参考基因组数据生成用于后续比对任务的索引,得到带有索引的参考基因组数据;步骤1)中根据预设选择对参考基因组数据、参考转录组数据进行预处理时,如果需要预处理参考转录组数据,则对参考转录组数据进行预处理的详细步骤包括:读取参考转录组数据,并通过cpu调用基于fpga上硬件实现的索引生成器为参考转录组数据生成用于后续比对任务的索引,得到带有索引的参考转录组数据;步骤1)中对原始的测序样本数据进行预处理的详细步骤包括:读取原始的测序样本数据,所述原始的测序样本数据进行数据质量控制,得到可靠测序样本数据;
优选地,所述数据质量控制包括移除reads上的接头序列、低质量序列、污染物和人造物。
优选地,步骤1)中对参考基因组数据进行预处理、对参考转录组数据进行预处理、对原始的测序样本数据进行预处理分别采用不同的线程或进程并发执行。
优选地,步骤2)中将可靠测序样本数据和参考基因组数据进行比对的详细步骤包括:
2.1a)读取可靠测序样本数据、读取预处理后的参考基因组数据及其索引;
2.2a)根据参考基因组数据的索引,基于cpu调用fpga上硬件实现的比对器将可靠测序样本数据和参考基因组数据进行比对,建立可靠测序样本数据和参考基因组数据之间的映射关系;
2.3a)根据比对结果移除重复的reads;
2.4a)输出可靠测序样本数据和参考基因组数据的比对结果信息。
优选地,步骤2)中将可靠测序样本数据和参考转录组数据进行比对的详细步骤包括:
2.1b)读取可靠测序样本数据、读取预处理后的参考转录组数据及其索引;
2.2b)根据参考转录组数据的索引,基于cpu调用fpga上硬件实现的比对器将可靠测序样本数据和参考转录组数据进行比对,建立可靠测序样本数据和参考转录组数据之间的映射关系;
2.3b)根据比对结果移除重复的reads;
2.4b)输出可靠测序样本数据和参考转录组数据的比对结果信息。
优选地,步骤3)中将可靠测序样本数据重新装配成转录本的详细步骤包括:
3.1a)读取可靠测序样本数据;
3.2a)基于cpu调用fpga上硬件实现的装配器将可靠测序样本数据重新装配成转录本,并建立上述转录组测序可靠样本数据和新转录本之间的映射关系;
3.3a)在转录本水平、基因水平、外显子水平三者中的至少一种对上述重新装配成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;
3.4a)输出重新装配的转录本及其量化结果信息。
优选地,步骤3)中对可靠测序样本数据和参考基因组数据及其比对结果进行转录本识别的详细步骤包括:
3.1b)读取可靠测序样本数据和参考基因组数据及其比对结果;
3.2b)判断是否提供已知参考模型文件,所述已知参考模型文件用于描述参考基因组上已知的包括基因、转录本、外显子、内含子在内的位置和属性;如果提供已知参考模型文件,则跳转执行步骤3.3b);否则,跳转执行步骤3.5b);
3.3b)根据对可靠测序样本数据和参考基因组数据及其比对结果进行转录本识别,识别并重构可靠测序样本数据的转录本;
3.4b)在转录本水平、基因水平、外显子水平三者中的至少一种对步骤3.3b)识别并重构成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;跳转执行步骤3.7b);
3.5b)根据可靠测序样本数据和参考基因组数据及其比对结果,发现并重构上述转录组测序可靠样本数据的转录本;
3.6b)在转录本水平、基因水平、外显子水平三者中的至少一种对步骤3.5b)识别并重构成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;跳转执行步骤3.b7);
3.7b)在所有转录本精确量化完毕后,进行转录本合并和再量化;
3.8b)基于cpu调用gpu上编程实现的识别器对合并和再量化后的转录本进行识别差异表达基因和差异表达转录本;
3.9b)输出转录本识别和量化结果信息。
优选地,步骤3)中对可靠测序样本数据和参考转录组数据及其比对结果进行转录本识别的详细步骤包括:
3.1c)读取可靠测序样本数据和参考转录组数据及其比对结果;
3.2c)根据可靠测序样本数据和参考转录组数据及其比对结果,识别并重构可靠测序样本数据的转录本;
3.3c)在转录本水平、基因水平、外显子水平三者中的至少一种对步骤3.2c)识别并重构成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;
3.4c)在所有转录本精确量化完毕后,进行转录本合并和再量化;
3.5c)基于cpu调用gpu上编程实现的识别器对合并和再量化后的转录本进行识别差异表达基因和差异表达转录本;
3.6c)输出转录本识别和量化结果信息。
优选地,步骤4)的详细步骤包括:
4.1)读取步骤3)输出的所有转录本识别和量化结果信息;
4.2)基于cpu调用gpu和dsp将所有转录本识别和量化结果信息以预设的可视化表现形式生成可视化结果,其中gpu上编程处理视频、动画和显示任务,dsp上编程处理图形、图像和音频任务;
4.3)针对将所有转录本识别和量化结果信息以预设的可视化表现形式生成可视化结果,通过cpu调用fpga上硬件实现的深度学习模型并行执行分析和挖掘,从而得到对应的数据挖掘和分析结果并输出。
本发明的转录组测序数据计算解读方法具有下述优点:
1、本发明的转录组测序数据计算解读方法对于转录组测序数据计算解读流程中的每个耗时瓶颈,基于任务本身的算法或模型并行性,结合cpu、fpga、gpu和dsp这四种处理器的特点,分别进行了有针对性的并行加速,提高了转录组测序数据计算解读的实时性。
2、本发明的转录组测序数据计算解读方法对于转录组测序数据计算解读流程中的转录组识别和转录组功能分析与挖掘,基于任务本身的目标,结合cpu、fpga、gpu和dsp这四种处理器的特点,引入了深度学习模型,加快和丰富了深度学习源数据的处理,提高了转录组测序数据计算解读的深度和广度。
3、本发明的转录组测序数据计算解读方法对于转录组测序数据计算解读流程中的数据可视化,结合cpu、gpu和dsp这三种处理器的特点,配合完成可视化处理,提高了转录组测序数据可视化的实时性,丰富了转录组测序数据可视化的多样性。
附图说明
图1为本发明实施例方法的基本流程示意图。
图2为本发明实施例方法的预处理流程示意图。
图3为本发明实施例方法的数据比对流程示意图。
图4为本发明实施例方法的转录组装配和识别流程示意图。
图5为本发明实施例方法的数据可视化和转录组功能分析挖掘流程示意图。
具体实施方式
如图1所示,本实施例的转录组测序数据计算解读方法的步骤包括:
1)输入参考基因组数据、参考转录组数据和原始的测序样本数据,根据预设选择通过cpu调用基于fpga上硬件实现的索引生成器对参考基因组数据、参考转录组数据进行预处理,对原始的测序样本数据进行预处理得到可靠测序样本数据;这个步骤需要使用cpu和fpga这两种处理器;
2)选择执行基于cpu调用fpga上硬件实现的比对器执行将可靠测序样本数据和参考基因组数据进行比对(alignment)、将可靠测序样本数据和参考转录组数据进行比对(alignment)两种任务;这个步骤需要使用cpu和fpga这两种处理器;
3)基于cpu调用fpga上硬件实现的装配器将可靠测序样本数据重新装配(denovoassembly)成转录本,选择执行基于cpu调用gpu上编程实现的识别器1对可靠测序样本数据和参考基因组数据及其比对结果进行转录本识别、基于cpu调用gpu上编程实现的识别器2对可靠测序样本数据和参考转录组数据及其比对结果进行转录本识别;在选择全部执行的情况下,这个步骤需要使用cpu、fpga和gpu这三种处理器;
4)基于cpu调用gpu和dsp将转录组的识别或重新装配的处理结果进行可视化处理,通过cpu调用fpga上硬件实现的深度学习(deeplearning,dl)模型在可视化处理得到的结果的基础上进行指定的数据挖掘和分析。这个步骤需要使用cpu、fpga、gpu和dsp这四种处理器。
参见图1,步骤1)和2)完成转录组测序数据的计算任务;步骤3)和4)完成转录组测序数据的解读任务。以下详细步骤描述中,如果未加特殊说明,缺省使用的是cpu。
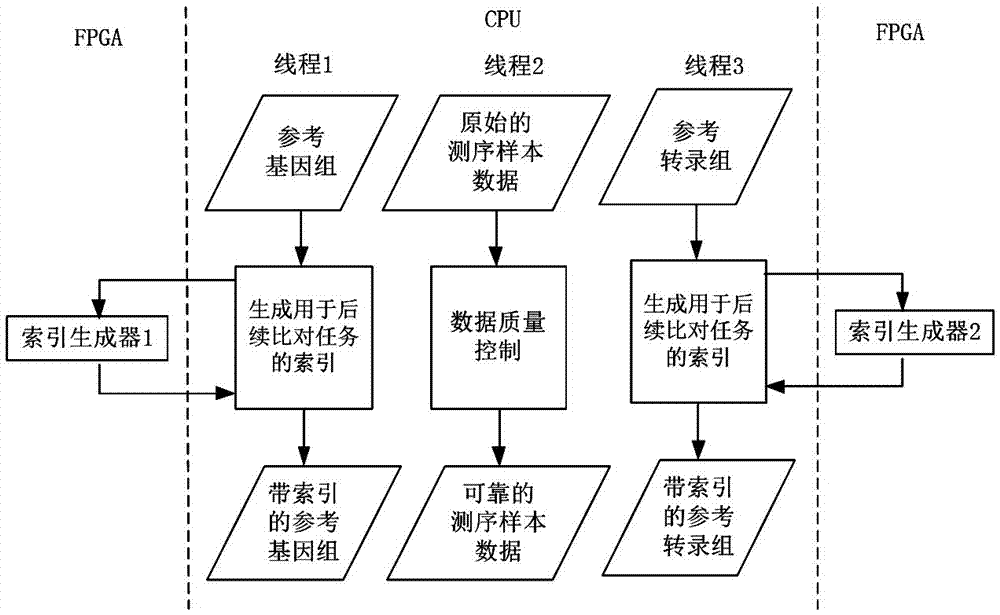
如图2所示,步骤1)中根据预设选择对参考基因组数据、参考转录组数据进行预处理时,如果需要预处理参考基因组数据,则对参考基因组数据进行预处理的详细步骤包括:读取参考基因组数据,并通过cpu调用基于fpga上硬件实现的索引生成器1为参考基因组数据生成用于后续比对任务的索引,得到带有索引的参考基因组数据。为参考基因组数据生成用于后续比对任务的索引时,cpu负责索引生成的流程控制,fpga上硬件实现的索引生成器1负责并行生成索引,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入fpga,能够并行加速完成其中的计算密集任务。虽然一段时间内,特定的参考基因组数据相对固定,可以生成索引一次,再在同类应用中反复使用,但是,一旦参考基因组数据有了更新,必须重新生成新索引。
如图2所示,步骤1)中根据预设选择对参考基因组数据、参考转录组数据进行预处理时,如果需要预处理参考转录组数据,则对参考转录组数据进行预处理的详细步骤包括:读取参考转录组数据,并通过cpu调用基于fpga上硬件实现的索引生成器2为参考转录组数据生成用于后续比对任务的索引,得到带有索引的参考转录组数据。为参考转录组数据生成用于后续比对任务的索引时,cpu负责索引生成的流程控制,fpga上硬件实现的索引生成器2负责并行生成索引,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入fpga,能够并行加速完成其中的计算密集任务。虽然一段时间内,特定的参考转录组数据相对固定,可以生成索引一次,再在同类应用中反复使用,但是,一旦参考转录组数据有了更新,必须重新生成新索引。
如图2所示,步骤1)中对原始的测序样本数据进行预处理的详细步骤包括:读取原始的测序样本数据,所述原始的测序样本数据进行数据质量控制,得到可靠测序样本数据(cleandatas);所述数据质量控制包括移除reads上的接头序列(theadaptersequences)、低质量序列(low-qualitysequences)、污染物(contaminants)和人造物(artifacts)。
本实施例中,步骤1)中对参考基因组数据进行预处理、对参考转录组数据进行预处理、对原始的测序样本数据进行预处理分别采用不同的线程或进程并发执行。参见图2,步骤1)包括3个并发执行的子任务:参考基因组的预处理、参考转录组的预处理和转录组测序原始样本数据的预处理。根据需要,在实际的转录组测序数据计算解读流程中,对于参考基因组的预处理和参考转录组的预处理,若想最大化利用已知的转录组研究领域的成果并且有2种参考数据,就都做;若只需或只有其一的参考数据,就只做其中之一;若没有参考数据或想发现新的转录本,可以都不做。
如图3所示,步骤2)包括2个并发执行的子任务:上述转录组测序可靠样本数据和上述参考基因组数据及其索引进行比对;上述转录组测序可靠样本数据和上述参考转录组数据及其索引进行比对。根据需要,在实际的转录组测序数据计算解读流程中,对于这2个子任务,若想最大化利用已知的转录组研究领域的成果并且有2种参考数据,就都做;若只需或只有其一的参考数据,就只做其中之一;若没有参考数据或想发现新的转录本,可以都不做。
步骤2)中将可靠测序样本数据和参考基因组数据进行比对的详细步骤包括:
2.1a)读取可靠测序样本数据、读取预处理后的参考基因组数据及其索引;
2.2a)根据参考基因组数据的索引,基于cpu调用fpga上硬件实现的比对器1将可靠测序样本数据和参考基因组数据进行比对,建立可靠测序样本数据和参考基因组数据之间的映射关系;将上述转录组测序可靠样本数据和上述参考基因组数据进行比对时,cpu负责数据比对的流程控制,fpga上硬件实现的比对器1负责并行执行数据比对,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入fpga,能够并行加速完成其中的计算密集任务。
2.3a)根据比对结果移除重复(duplicate)的reads;
2.4a)输出可靠测序样本数据和参考基因组数据的比对结果信息。
如图3所示,步骤2)中将可靠测序样本数据和参考转录组数据进行比对的详细步骤包括:
2.1b)读取可靠测序样本数据、读取预处理后的参考转录组数据及其索引;
2.2b)根据参考转录组数据的索引,基于cpu调用fpga上硬件实现的比对器2将可靠测序样本数据和参考转录组数据进行比对,建立可靠测序样本数据和参考转录组数据之间的映射关系;将上述转录组测序可靠样本数据和上述参考转录组数据进行比对时,cpu负责数据比对的流程控制,fpga上硬件实现的比对器2负责并行执行数据比对,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入fpga,能够并行加速完成其中的计算密集任务。
2.3b)根据比对结果移除重复(duplicate)的reads;
2.4b)输出可靠测序样本数据和参考转录组数据的比对结果信息。
本实施例中,步骤3)包括3个并发执行的子任务:上述转录组测序可靠样本数据重新装配成转录本;基于上述转录组测序可靠样本数据和上述参考基因组数据及其索引比对结果的转录本识别;基于上述转录组测序可靠样本数据和上述参考转录组数据及其索引比对结果的转录本识别。根据需要,在实际的转录组测序数据计算解读流程中,对于这3个子任务,若想最大化利用已知的转录组研究领域的成果并且有2种参考数据,就都做;若只需或只有其一的参考数据,就选做基于上述转录组测序可靠样本数据和上述参考基因组数据及其索引比对结果的转录本识别及基于上述转录组测序可靠样本数据和上述参考转录组数据及其索引比对结果的转录本识别其中之一;若没有参考数据或想发现新的转录本,就只做或加做上述转录组测序可靠样本数据重新装配成转录本。
如图4所示,步骤3)中将可靠测序样本数据重新装配成转录本的详细步骤包括:
3.1a)读取可靠测序样本数据;
3.2a)基于cpu调用fpga上硬件实现的装配器将可靠测序样本数据重新装配成转录本,并建立上述转录组测序可靠样本数据和新转录本之间的映射关系;将上述转录组测序可靠样本数据重新装配成转录本时,cpu负责转录本重新装配的流程控制,fpga上硬件实现的装配器负责并行执行数据装配,cpu和fpga之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入fpga,能够并行加速完成其中的计算密集任务。
3.3a)在转录本水平、基因水平、外显子水平三者中的至少一种对上述重新装配成的转录本的基本特性进行精确量化(quantification),所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;本实施例中,可根据需要,在转录本水平(transcriptlevel),以及/或者基因水平(genelevel),以及/或者外显子水平(exonlevel),对上述装配转录本的基本特性,例如表示可靠样本数据的丰度(abundant)的fpkm(fragmentsperkilobasemillion)、rpkm(readsperkilobasemillion)、tpm(transcriptsperkilobasemillion),外显子-内含子结构(exon-intronstructure),及相关基因和亚型(isoforms)等,进行精确量化(quantification);
3.4a)输出重新装配的转录本及其量化结果信息。
如图4所示,步骤3)中对可靠测序样本数据和参考基因组数据及其比对结果进行转录本识别的详细步骤包括:
3.1b)读取可靠测序样本数据和参考基因组数据及其比对结果;
3.2b)判断是否提供已知参考模型文件,所述已知参考模型文件用于描述参考基因组上已知的包括基因、转录本、外显子、内含子在内的位置和属性,可以引导转录本的识别,而且也有助于低丰度(low-abundance)基因的重构(reconstruction);如果提供已知参考模型文件,则跳转执行步骤3.3b);否则,跳转执行步骤3.5b);
3.3b)根据对可靠测序样本数据和参考基因组数据及其比对结果进行转录本识别,识别并重构可靠测序样本数据的转录本;
3.4b)在转录本水平、基因水平、外显子水平三者中的至少一种对步骤3.3b)识别并重构成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;跳转执行步骤3.7b);可根据需要,在转录本水平,以及/或者基因水平,以及/或者外显子水平,对上述的识别转录本的基本特性,如表示可靠样本数据丰度的fpkm、rpkm、tpm,外显子-内含子结构,相关基因和亚型,以及和已知参考模型文件中已知基因、转录本、外显子-内含子结构的匹配性,全新基因、转录本、外显子-内含子结构的数量等,进行精确量化;
3.5b)根据可靠测序样本数据和参考基因组数据及其比对结果,发现并重构上述转录组测序可靠样本数据的转录本;
3.6b)在转录本水平、基因水平、外显子水平三者中的至少一种对步骤3.5b)识别并重构成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;跳转执行步骤3.b7);可根据需要,在转录本水平,以及/或者基因水平,以及/或者外显子水平,对上述的发现转录本的基本特性,如表示可靠样本数据丰度的fpkm、rpkm、tpm,外显子-内含子结构,相关基因和亚型等,进行精确量化;
3.7b)在所有转录本精确量化完毕后,进行转录本合并和再量化;
3.8b)基于cpu调用gpu上编程实现的识别器1对合并和再量化后的转录本进行识别差异表达基因和差异表达转录本;识别差异表达基因(differentiallyexpressedgenes,degs)和差异表达转录本(differentiallyexpressedtranscripts,dets),包括上述识别或发现转录本和上述参考基因组之间,以及多个测序样本对应的识别或发现转录本间的差异表达识别。其中,cpu负责差异表达识别的流程控制,gpu上编程实现的识别器1负责并行执行差异表达识别,cpu和gpu之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入gpu,能够并行加速完成其中的计算密集任务。
3.9b)输出转录本识别和量化结果信息。
如图4所示,步骤3)中对可靠测序样本数据和参考转录组数据及其比对结果进行转录本识别的详细步骤包括:
3.1c)读取可靠测序样本数据和参考转录组数据及其比对结果;
3.2c)根据可靠测序样本数据和参考转录组数据及其比对结果,识别并重构可靠测序样本数据的转录本;
3.3c)在转录本水平、基因水平、外显子水平三者中的至少一种对步骤3.2c)识别并重构成的转录本的基本特性进行精确量化,所述基本特性包括表示可靠样本数据的丰度的fpkm、rpkm、tpm、外显子-内含子结构及相关基因、亚型中的至少一种;可根据需要,在转录本水平,以及/或者基因水平,以及/或者外显子水平,对上述的识别转录本的基本特性,如表示可靠样本数据丰度的fpkm、rpkm、tpm,外显子-内含子结构,相关基因和亚型等,进行精确量化;
3.4c)在所有转录本精确量化完毕后,进行转录本合并和再量化;
3.5c)基于cpu调用gpu上编程实现的识别器2对合并和再量化后的转录本进行识别差异表达基因和差异表达转录本;识别差异表达基因(differentiallyexpressedgenes,degs)和差异表达转录本(differentiallyexpressedtranscripts,dets),包括上述识别或发现转录本和上述参考转录组之间,以及多个测序样本对应的识别或发现转录本间的差异表达识别。其中,cpu负责差异表达识别的流程控制,gpu上编程实现的识别器2负责并行执行差异表达识别,cpu和gpu之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入gpu,能够并行加速完成其中的计算密集任务。
3.6c)输出转录本识别和量化结果信息。
如图5所示,步骤4)的详细步骤包括:
4.1)读取步骤3)输出的所有转录本识别和量化结果信息;分别包括读取上述步骤3.4a)重新装配的转录本及其量化结果信息#1;读取上述步骤3.9b)转录本识别和量化结果信息#2;读取上述步骤3.6c)转录本识别和量化结果信息#3;
4.2)基于cpu调用gpu和dsp将所有转录本识别和量化结果信息以预设的可视化表现形式生成可视化结果,其中gpu上编程处理视频、动画和显示任务,dsp上编程处理图形、图像和音频任务;将上述重新装配的转录本及其量化结果信息#1、转录本识别和量化结果信息#2和转录本识别和量化结果信息#3进行可视化,即以各种科学、直观、生动的方式表现数据的含义。例如:将上述fpkm、rpkm、tpm,表示外显子-内含子结构,相关基因和亚型等的量化值进行图、表、形、动画、音频、视频等等的可视化处理。其中,cpu负责可视化的流程控制;gpu上编程处理视频、动画和显示等任务,cpu和gpu之间有数据和指令交互;dsp上编程处理图形、图像和音频等任务,cpu和dsp之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入gpu和dsp,它们和cpu相互配合,能够并行加速完成多媒体处理任务。
4.3)针对将所有转录本识别和量化结果信息以预设的可视化表现形式生成可视化结果,通过cpu调用fpga上硬件实现的深度学习模型并行执行分析和挖掘,从而得到对应的数据挖掘和分析结果并输出。本实施例中通过转录组功能分析和挖掘,即基于上述分析数据,进一步深入分析相关的转录组功能,例如:基因边界鉴定、可变剪切等的转录本结构研究;基因融合、编码区单核苷酸多态性(codingsinglenucleotidepolymorphism,csnp)等的转录本变异研究;ncrna、microrna等的非编码区域功能研究。并扩大外延,在已知的知识之外,再挖掘未知的关联。例如:发现全新(denovo)转录本和稀有转录本,分析、挖掘、预测和鉴定它们的作用靶基因,转录体聚类和表达谱分析等。其中,cpu负责分析和挖掘的流程控制;fpga上硬件实现的深度学习模型负责并行执行分析和挖掘,cpu和fpga之间有数据和指令交互。使用深度学习方法进行分析和挖掘,可以支持基于大数据的统计学模型,能够实现更加准确的分析和挖掘;gpu上编程处理分析挖掘相关的视频、动画和显示等任务,cpu和gpu之间有数据和指令交互;dsp上编程处理和分析挖掘相关的图形、图像和音频等任务,cpu和dsp之间有数据和指令交互。只使用cpu时,这一步骤是整个转录组测序数据计算解读流程中的耗时瓶颈之一,加入fpga、gpu和dsp,它们和cpu相互配合,能够并行加速完成深度学习及相关多媒体处理任务;
综上所述,本实施例的转录组测序数据计算解读方法能够满足测序数据计算解读的快速实时、精准深入、通俗易懂、形式多样的要求,为转录组测序技术的应用推广助力。
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!