一种基于基因表达谱的胃癌预后标志物筛选及分类方法与流程
本发明涉及生物研究
技术领域:
,具体为一种基于基因表达谱的胃癌预后标志物筛选及分类方法。
背景技术:
:胃癌是最常见的恶性肿瘤之一,发病率及死亡率居高不下,早期诊断、合理评估其预后并适时适度干预十分重要。目前研究揭示:胃癌是一种基因病,是由多种癌基因抗癌基因共同参与、多阶段多途径协同,使胃黏膜逐步发展到癌前病变,再发展到胃癌的这样一个演变过程。正常胃黏膜到癌前变过程及癌前变至胃癌过程皆存在特征性的差异表达基因,而分子病理学将分子杂交技术与组织形态学检测相融合,随着免疫组织化学及分子生物学的发展,胃癌的病理与免疫学及分子生物学研究之间越来越密不可分,目前已探索了许多与胃癌相关的基因及其蛋白质产物,早期发现这些基因及标志物为探讨胃癌发病机理开辟了新途径,更加能够揭示肿瘤组织的生长活性在肿瘤生长、浸润和转移方面的作用,从而更加准确地指导临床治疗,判断预后。随着基因芯片实验技术的日益成熟和完善,各种基因表达谱数据正在以指数级方式增加。目前,全球最有影响力的基因表达谱数据库主要包括GEO(全称是GeneExpressionOmnibus)、ArrayExpress以及SMD(全称是StanfordMicroarrayDatabase)。生物信息技术也在快速发展,利用基因表达谱数据在基因水平上研究肿瘤的发生发展机理,有助于肿瘤诊断和个性化治疗。当前基于基因表达谱的肿瘤分类方法研究多数集中在两方面:一、由于基因表达谱数据具有高维样本的显著特性,而且其中存在大量的冗余基因及噪声,如何从高维数据中提取出致病基因仍是一个难点;二、基因表达谱数据分析的准确度还没有达到应用水平,肿瘤的最终诊断例如胃癌等,还是依赖于医学专家。因此寻找合适的分类算法并提高其性能是目前研究的重中之重。技术实现要素:本发明的目的在于提供一种基于基因表达谱的胃癌预后标志物筛选及分类方法,以解决上述
背景技术:
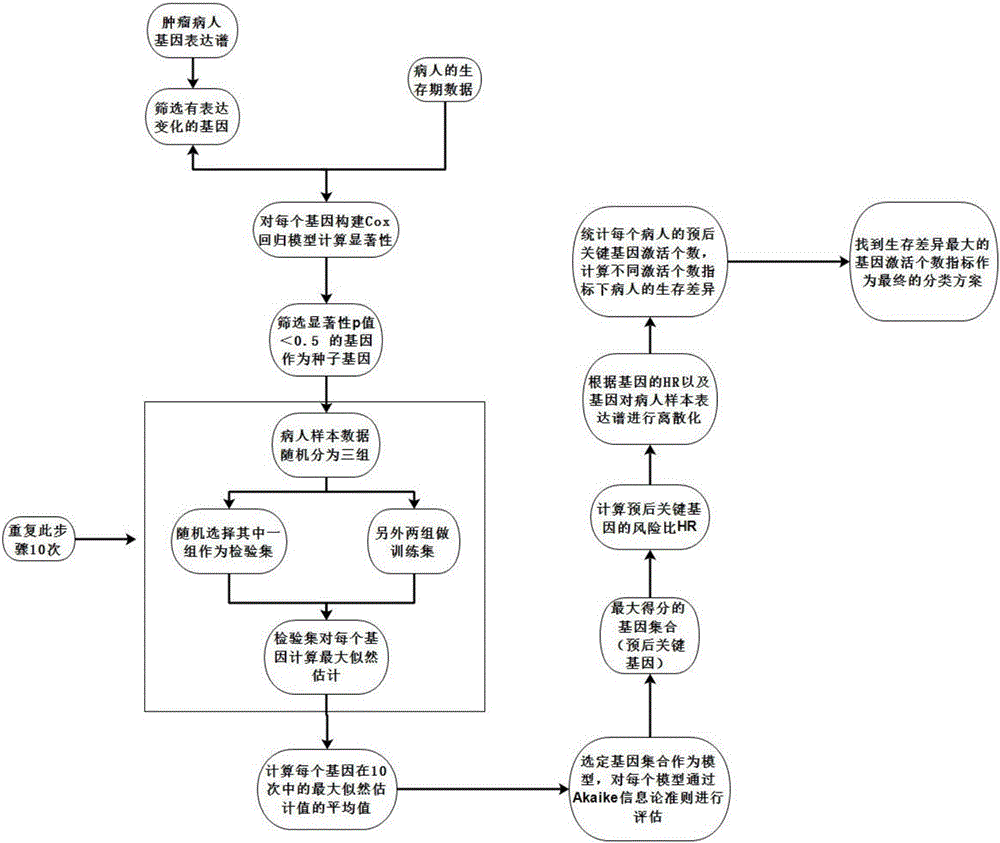
中提出的问题。为了实现上述目的,本发明一种基于基因表达谱的胃癌预后标志物筛选及分类方法,包括以下步骤(1)从GEO数据库获取胃癌病人基因表达谱数据以及病人的临床随访信息数据,且数据样本的数目为N。(2)根据步骤(1)中得到的基因表达谱数据构建病人的基因表达谱矩阵,若出现某个基因在某个样本中未检测到则使用该基因在其他样本中的表达平均值替代,若出现多次检测到则取平均值替代,最终构建无缺失值的矩阵如下表所列:Sample1Sample2……SampleNGene1Exp11Exp12Exp1…Exp1NGene2Exp21Exp22Exp2…Exp2N……Exp…1Exp…2Exp……Exp…NGeneMExpM1ExpM2ExpM…ExpMN(3)根据步骤(2)所得矩阵计算每个基因(i=1,2,3……M)在各样本中的表达水平的中位数Mi,以及在各样本中表达水平的方差Vi,进一步的将所有基因的中位数Mi和方差Vi从大到小排序,分别选取在排序前80%的基因组成两个基因集合,进一步的选取两个基因集合取交集作为预选的有变化的基因。(4)根据步骤(3)中筛选出来的基因的表达水平以及步骤(1)中的病人的临床随访信息数据建立Cox比例风险模型:其中β1,β2,…,βp为偏回归系数,h0(t)为未知数,x1,x2,…,xp为基因的表达水平,h(t)表示t时刻病人死亡的风险率。对Cox模型检验,采用似然比检验,步骤如下:a、假设H0:所有的βi为0,H1:所有的βi不为0;b、将H0和H1条件下的最大似然函数的对数值分别记为LLp(H0)和LLp(H1);c、计算在原假设的条件下统计量χ2=-2[LLp(H1)-LLp(H0)]服从自由度为p的χ2分布的显著性p值;最后根据上述a、b、c步骤对每一个基因单独代入Cox模型,分别计算最终的统计学显著性p值,最终筛选显著性p值小于0.05的基因作为种子基因。(5)根据种子基因在病人中表达水平构建新的表达矩阵,结合步骤(1)中的病人的临床随访信息数据构建最大似然模型筛选预后关键基因,步骤如下:Ⅰ、随机将样本分成三份,选择三倍交叉验证,随机选择其中两组作为训练集,另外一组作为检验集。根据训练集可以得到每个基因的参数的估计值,然后这个参数在检验集通过最大似然估计方法评价好坏。Ⅱ、重复第Ⅰ步10次,得到每一个基因的最大似然估计的10个值,选择最大似然估计平均值最大的作为最好的基因。接着搜索下一个最好的基因,评估剩下的每个基因与上次最好的基因的组合模型。Ⅲ、通过不断的向前选择基因的方法得到一系列模型。对每个模型通过Akaike信息论准则(AICs)进行模型的评估,最后选择AIC值最小的那个模型作为最优模型,筛选出最优的基因组合作为预后关键基因。Ⅳ、计算基因i的风险比HR,公式如下:(6)根据步骤(5)中筛选出来的每个预后关键基因在病人中的风险比以及这些基因对应病人的表达谱构建新的表达矩阵,然后对矩阵进行离散化,离散方式如下:当预后关键基因i对应风险系数大于1且该基因在样本j中的表达水平排在该基因在所有样本中的表达水平的中位数以上,则在矩阵中将表达水平替换成1。当预后关键基因i对应风险系数小于1且该基因在样本j中的表达水平排在该基因在所有样本中的表达水平的中位数以下,则在矩阵中将表达水平替换成1。不满足以上两个条件的则标记为0。最后得到0-1矩阵。(7)根据病人预后关键基因的0-1矩阵,统计每个病人在各预后关键基因中被标记为1的个数Ci,分别根据Ci≥1,Ci≥2…..Ci≥N(N=预后关键基因个数)对病人进行高风险预测归类。(8)根据步骤(7)中N种归类方式,使用Kaplan-Meier生存函数估计预测为高风险病人与其他病人的生存差异,采用log-rank检验方式作为统计学检验特征,最终得到每个归类方式下的显著性p值,选择最显著的归类方式作为最终的归类方式对病人进行高风险预测。优选的,所述步骤(1)中的病人基因表达谱数据以及病人的临床随访信息数据的数据样本量N至少为11。优选的,从所述步骤(4)中筛选的显著性p值小于0.05的基因中筛选p值较小的部分基因作为种子基因。与现有技术相比,本发明的有益效果是:一种基于基因表达谱的胃癌预后标志物筛选及分类方法,采用多种特征提取的方法组合将高维表达谱数据降维挖掘出最重要的几个影响疾病的关键的基因,大大降低基因的维数,从而提高分类正确率,并根据挖掘得到的疾病预后关键基因的表达水平使用离散化方法预测病人的预后风险。附图说明图1为本发明的流程示意图。具体实施方式下面结合具体实施例对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。一种基于基因表达谱的胃癌预后标志物筛选及分类方法:从GEO数据库中下载432个胃癌的疾病样本一共17418个基因的表达值以及病人的病人的临床随访数据,并建立数据矩阵。筛选在各样本中有变化的基因,计算每个基因(i=1,2,3……M)在各样本中的表达水平的中位数Mi,以及在各样本中表达水平的方差Vi,进一步的将所有基因的中位数Mi和方差Vi从大到小排序,分别选取在排序前80%的基因组成两个基因集合,进一步的选取两个基因集合取交集作为预选的有变化的基因。通过此筛选条件从17418个基因中共得到了11420个有变化的基因。根据基因在各病人中的表达水平以及对应的病人的生存时间借助于Logistic模型的构造思想构建Cox比例风险模型:其中β1,β2,…,βp为偏回归系数,h0(t)为未知数,x1,x2,…,xp为基因的表达水平,h(t)表示t时刻病人死亡的风险率。对Cox模型检验,采用似然比检验,步骤如下:a、假设H0:所有的βi为0,H1:所有的βi不为0;b、将H0和H1条件下的最大似然函数的对数值分别记为LLp(H0)和LLp(H1);c、计算在原假设的条件下统计量χ2=-2[LLp(H1)-LLp(H0)]服从自由度为p的χ2分布的显著性p值;最后根据上述步骤对每一个基因单独代入以上模型,分别计算最终的统计学显著性p值,得到共有798个显著性p值小于0.05的基因,进一步筛选出其中最显著的20个基因如下表所列:Ⅰ、选择三倍交叉验证(随机分成三份),随机将每一个样本分配到训练集和检验集。根据训练集得到每个基因的参数的估计值,然后这个参数在检验集通过最大似然估计方法评价好坏。Ⅱ、重复步骤Ⅰ这一步10次,得到每一个基因的最大似然估计的10个值,选择最大似然估计平均值最大的作为最好的基因。接着搜索下一个最好的基因,评估剩下的每个基因与上次最好的基因的组合模型。Ⅲ、通过不断的向前选择基因的方法得到一系列模型。对每个模型通过Akaike信息论准则(AICs)进行模型的评估,最后选择AIC值最小的那个模型作为最优模型。Ⅳ、重复以上步骤Ⅰ、步骤Ⅱ和步骤Ⅲ循环1000次,结果显示9个基因的组合频率为999次,选取这9个基因作为预后关键基因;同时计算出风险比HR,其计算公式为根据步骤Ⅳ筛选出来的预后关键基因在病人中的风险比以及这些预后关键基因对应病人的表达谱构建新的表达矩阵,然后对矩阵进行离散化,离散方式如下:当预后关键基因i对应风险系数大于1且该基因在样本j中的表达水平排在该基因在所有样本中的表达水平的中位数以上,则在矩阵中将表达水平替换成1。当关键基因i对应风险系数小于1且该基因在样本j中的表达水平排在该基因在所有样本中的表达水平的中位数以下,则在矩阵中将表达水平替换成1。不满足以上两个条件的则标记为0,计算出每个样本对应的激活影响因子个数。根据每个样本对应的激活影响因子个数≥1、≥2、≥3、≥4……进行分类,同时进行KaplanMeier单因素生存分析得到结果显著性p值,不同分类方式得到的KaplanMeier生存分析结果如下表所列:激活影响因子个数显著性p值激活影响因子个数≥10.2052激活影响因子个数≥20.00264激活影响因子个数≥36e-05激活影响因子个数≥43.108872e-08激活影响因子个数≥53.830298e-10激活影响因子个数≥61.841547e-08激活影响因子个数≥74.573210e-10激活影响因子个数≥83.751351e-08激活影响因子个数≥91.216125e-05从表中可以看出整体九个分类结果都有着显著的预后差异,尤其激活影响因子个数≥5最为显著,最终我们选择激活影响因子个数≥5作为最终的模型,即九个预后特征基因中有五个被激活则病人具有预后高风险。使用TCGA引入外部数据,对以上结果进行验证,根据激活影响因子个数≥5将415个病人样本分为高风险病人和低风险病人两组,使用KaplanMeier单因素生存分析结果显示两类样本的生存时间有显著性p值为0.00445,复发风险的显著性p值为0.00147,具有显著差别。本发明的基于因表达谱数据的胃癌预后标志物筛选及分类方法可以有效的将高维冗杂的基因表达谱数据降维,从几万个基因中筛选出容易应用于临床检测的关键的几个基因;并且通过本发明提供的分类方法可以通过其他实验室的数据得到验证。尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1