固态电池
固态电池
发明领域
1.本发明涉及具有碱金属硫化物固态电解质的固态电池的领域。
背景技术:
2.固态锂离子导体,实现所有固态锂离子电池的关键组件,是电池领域中最追求的研究目标之一。对固态电解质和更一般地对固态电池的强烈兴趣主要源于改进的安全性、实现新电极材料的能力和更好的低温性能。由于目前使用的液态电解质通常是高度易燃的有机溶剂,因此期望固态电池单元的安全性改进。用不易燃的固体代替这些电解质将消除电池安全性的最成问题的方面。此外,固态电解质与若干不能用基于液态电解质的配置实施的高能量密度电极材料相容。固态电解质还比液态电解质维持更好的低温操作,液态电解质在低温下经历显著的离子传导率下降。这种低温性能在快速增长的电动车辆市场中是关键的。
3.在目前研究的固态电解质中,硫化物仍然是最高性能和最有前途的家族之一。硫化物玻璃固态电解质和玻璃
‑
陶瓷固态电解质(其中晶相已经沉淀在玻璃基质内)已经分别展示0.1
‑
1ms cm
‑1数量级和高于1ms cm
‑1的离子传导率。陶瓷
‑
硫化物电解质,最显著的是li
10
gep2s
12
(lgps)和li
10
sip2s
12
(lsps),是特别有前途的,因为它们维持极高的离子传导率。lgps是最初的固态电解质之一,达到可与液态电解质相当的离子传导率12ms cm
‑1,仅被lsps替代,lsps实现了25ms cm
‑1的惊人高的离子传导率。尽管具有这些有前景的电导率,但陶瓷
‑
硫化物家族受到窄稳定窗口的困扰。也就是说,lgps和lsps两者都倾向于在相对于锂金属低于大约1.7v的电压下还原或在高于大约2.1v下氧化。已经证明这种有限的稳定窗口是需要在大约0
‑
4v的电压范围内工作的电池单元的主要障碍。
4.因此,需要一种改进的固态电池,其结合了具有可控结构性质和表面化学的固态电解质。
技术实现要素:
5.我们已经开发了使用具有改进的循环性能的固态电解质的可再充电的固态电池。本文公开的可再充电的固态电池是有利的,因为固态电解质具有优越的电压稳定性和优异的电池循环性能。本发明的电池可以针对锂枝晶的形成而稳定化和/或可以在高电流密度下工作延长的循环次数。
6.在一个方面,本发明的特征在于可再充电的电池,其包括第一电极、第二电极和布置在其间的固态电解质。所述固态电解质包括硫化物,所述硫化物包含碱金属,例如锂。在某些实施方案中,固态电解质处于足以在电化学循环期间稳定固态电解质的体积约束下。在特定实施方案中,所述体积约束对固态电解质施加约70mpa至约1,000mpa,例如约100
‑
250mpa的压力,例如对固态电解质的显微结构施加15gpa数量级的机械压缩。在某些实施方案中,体积约束提供1v至10v,例如1
‑
8v、5.0
‑
8v或大于5.7v或甚至大于10v的电压稳定窗口。
7.在一些实施方案中,固态电解质具有核壳形态。在某些实施方案中,碱金属是li、na、k、rb或cs,例如li。在一些实施方案中,固态电解质包含sips、geps、snps、psi或ps。在一些实施方案中,固态电解质是li
10
sip2s
12
、li
10
gep2s
12
或li
9.54
si
1.74
p
1.44
s
11.7
cl
0.3
。在一些实施方案中,第一电极是阴极,其可以包含licoo2、lini
0.5
mn
1.5
o4、v li2copo4f、linipo4、li2ni(po4)f、limnf4、lifef4或lico
0.5
mn
1.5
o4。在某些实施方案中,第二电极是阳极,并且可以包括锂金属、锂化石墨或li4ti5o
12
。在特定实施方案中,体积约束提供约1gpa至约100gpa,例如约15gpa的对固态电解质的机械压缩。
8.在另一方面,本发明的特征在于可充电的电池,其包括第一电极、第二电极和布置在其间的固态电解质,其中第二电极是包含碱金属和石墨的阳极。在一些实施方案中,电池处于约70
‑
1000mpa,例如约100
‑
250mpa的压力下。在特定的实施方案中,碱金属和石墨形成复合材料。在一些实施方案中,碱金属是li、na、k、rb或cs,例如li。在一些实施方案中,固态电解质包含sips、geps、snps、psi或ps。在某些实施方案中,固态电解质是li
10
sip2s
12
、li
10
gep2s
12
或li
9.54
si
1.74
p
1.44
s
11.7
cl
0.3
。在特定的实施方案中,第一电极是阴极,并且可以包含licoo2、lini
0.5
mn
1.5
o4、vli2copo4f、linipo4、li2ni(po4)f、limnf4、lifef4或lico
0.5
mn
1.5
o4。在一些实施方案中,电池处于提供约1gpa至约100gpa,例如约15gpa的对固态电解质的机械压缩的外部应力下。
9.在另一方面,本发明的特征在于可再充电的电池,其包括第一电极、第二电极和布置在其间的固态电解质,其中所述固态电解质可以包括包含碱金属的硫化物;并且所述电池处于等容约束下。在一些实施方案中,所述等容约束通过在约3
‑
1000mpa,例如约100
‑
250mpa的压力下压缩固态电解质来提供。在某些实施方案中,碱金属是li、na、k、rb或cs,例如li。在一些实施方案中,固态电解质包含sips、geps、snps、psi或ps。在某些实施方案中,固态电解质是li
10
sip2s
12
、li
10
gep2s
12
或li
9.54
si
1.74
p
1.44
s
11.7
cl
0.3
。在特定的实施方案中,第一电极是阴极,并且可以包含licoo2、lini
0.5
mn
1.5
o4、v li2copo4f、linipo4、li2ni(po4)f、limnf4、lifef4或lico
0.5
mn
1.5
o4。在一些实施方案中,等容约束提供约1gpa至约100gpa,例如约15gpa的对固态电解质的机械压缩。
10.在另一方面,本发明的特征在于可再充电的电池,其具有第一电极、第二电极和布置在其间的固态电解质。所述固态电解质包括硫化物,所述硫化物包含碱金属,并且任选地具有核
‑
壳形态。第一电极包含界面稳定涂覆材料。在某些实施方案中,第一电极和第二电极独立地包含界面稳定涂覆材料。在某些实施方案中,第一电极和第二电极中的一个包含锂
‑
石墨复合材料。
11.在一些实施方案中,第一电极包含如本文,例如表1中所述的材料。在一些实施方案中,第一电极的涂覆材料是如本文所述的涂覆材料,例如linbo3、alf3、mgf2、al2o3、sio2、石墨,或表2中所述的涂覆材料。在某些实施方案中,碱金属是li、na、k、rb或cs,例如li。在一些实施方案中,固态电解质包含sips、geps、snps、psi或ps。在某些实施方案中,固态电解质是li
10
sip2s
12
、li
10
gep2s
12
或li
9.54
si
1.74
p
1.44
s
11.7
cl
0.3
。在一些实施方案中,第一电极是阴极,并且可以包含lic
o
o2、lini
0.5
mn
1.5
o4、v li2copo4f、linipo4、li2ni(po4)f、limnf4、lifef4或lico
0.5
mn
1.5
o4。在一些实施方案中,电池处于提供约1gpa至约100gpa,例如约15gpa的对固态电解质的机械压缩的外部应力下。在某些实施方案中,电池处于约70
‑
1000mpa,例如约100
‑
250mpa的压力下。
12.在另一方面,本发明的特征在于通过跨第一电极和第二电极施加电压并对本发明的可再充电的电池充电来储存能量的方法。在另一方面,本发明提供通过将负载连接到第一电极和第二电极并允许本发明的可再充电的电池放电来提供能量的方法。
附图说明
13.图1a
‑
1b:在不同压力下以液态(a)和固态(b)的lgps的循环伏安法(cv)测试。在液态电解质中测试具有比率90∶10的lgps/c薄膜((a)中的黑色曲线)。cv测试还通过在不同压力下代替液态电解质用lgps球粒进行,其为全固态cv。分解强度随着施加压力的增加而显著降低。在6t(420mpa)的合理低压下,在5.7v之前已经没有明显的分解峰(紫色曲线),这指示对电池单元层面施加外部压力或体积压缩可以拓宽固态电解质的电化学窗口。
14.图2a
‑
2b:licoo2(lco)
‑
li4ti5o
12
(lto)全固态全电池的容量(a)和循环性能(b)。由于lto的化学势为1.5v(相对于li),所以阴极侧的工作平台高于4v(相对于li)。
15.图3a
‑
3b:lini
0.5
mn
1.5
o4(lnmo)
‑
lto全固态全电池的容量(a)和循环性能(b)。由于lto的化学势为1.5v(相对于li),所以阴极侧的工作平台高于4.7v(相对于li)。
16.图4:用于6v和更高的全固态锂离子电池技术的高电压阴极候选物。图例标记为:f是氟化物,o是氧化物,p,o是磷酸盐,并且s,o是硫酸盐。这些高电压氟化物、氧化物、磷酸盐和硫酸盐的完整列表提供于表1中。市售licoo2(lco)和lmno被标记为星星。
17.图5a
‑
5b:(a)应变对lgps分解的影响的图示,其中x
d
是已经分解的lgps的分数。下部虚线表示当施加可忽略的压力时,原始lgps和任意一组衰减产物(d)的二元组合的gibbs能(g
o
(x
d
))(等压衰减,p≈0gpα)。实线显示当将机械约束施加到lgps时的gibbs。由于lgps在分解时趋于膨胀,所以当施加这样的机械约束时,应变gibbs(g
应变
)增加。在一些破裂点,表示为x
f
,系统的gibbs能超过使机械约束破裂所需的能量(上部虚线)。突出显示的路径是机械约束的lgps系统的建议的基态。如果则区域x
d
<x
f
是亚稳的。(b)在类“流体”系统和类“固体”系统的情况下,功微分的示意性表示。对于顶部的“类流体”系统,所述系统由于分解而不是施加应力而经历内部体积膨胀(“无应力”应变)。底部系统表示远离任意参考状态的弹性变形。
18.图6:平均场极限中lgps和lgpso(li
10
gep2s
11.5
o
0.5
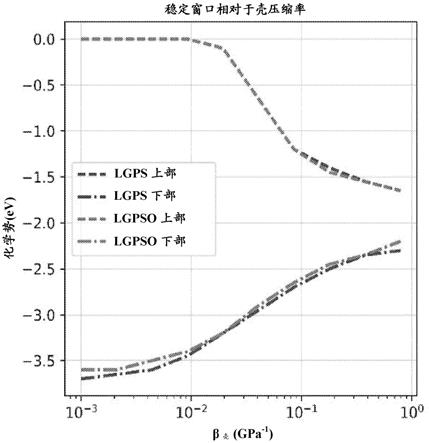
)的稳定窗口。β
壳
=v
核
‑1θ
p
v
核
指示约束机制的刚性程度。极限β
壳
→
0和β
壳
→
∞表示等容极限和等压极限。在等压的情况下,恢复了固有的材料稳定性(~1.7
‑
2.1v)。
19.图7a
‑
7b:(a)成核衰减机制的图示。半径r
o
的原始lgps颗粒在其中心处半径r
i
的区域内经历衰减。在不存在应力的情况下,分解的区域的半径现在为r
d
,其必定被挤入r
i
的空隙中。最终产生成核颗粒(iv),其中应变不是零。(b)流体静力学/平均场模型和成核模型两者的以kv为单位。对于典型的泊松比,可以看出应变项与理想的核
‑
壳模型(β
壳
=0)相当或比其更好。
20.图8a
‑
8e:在各种电池配置中的电压(φ)、锂化学势(μ
li
+)和费米能级(ε
f
)分布。(a)常规电池设计。(b)具有混合固态电解质/活性材料阴极的常规电池。χ
i
给出由于不同的锂离子化学势而在活性材料和固态电解质之间形成的界面电压。(c)绝缘层如何能够导致单元内的可变锂金属化学势的先前推测的图示。(d)考虑到由于锂空穴迁移而发生的有效
电子传导,预期来自部分(c)的电压如何驰豫。(e)施加的电压一旦超过固态电解质的固有稳定窗口,则部分(d)的结果。可以看出,在绝缘区域内形成局部锂,其中界面电压(χ
i
)等于施加的电压。
21.图9a
‑
9d:lgps和超lgps颗粒的显微结构和化学组成之间的比较。(a、c)分别为lgps和超lgps颗粒的典型tem明场图像,显示超lgps颗粒的明显表面层。(b、d)对不同尺寸的各种lgps和超lgps颗粒进行统计分析的stem eds线扫描,显示lgps颗粒的硫浓度从表面到本体均匀分布,而超lgps的硫浓度在表面层中降低。
22.图10:穿过具有100nm至3μm的不同粒度的单个lgps颗粒的stem eds线扫描,显示硫浓度从表面到本体的变化没有规则模式。
23.图11:穿过在碳酸二甲酯(dmc)中超声处理70小时的具有60nm至4μm范围的不同粒度的单个lgps颗粒的stem eds线扫描,显示与本体中的硫浓度相比,在表面区域的硫浓度明显较小。
24.图12:穿过在碳酸二乙酯(dec)中超声处理70小时的具有120nm至4μm范围的不同粒度的单个lgps颗粒的stem eds线扫描,显示与本体中的硫浓度相比,在表面区域的硫浓度明显较小。
25.图13:超声制备之前和之后lgps颗粒的定量stem edx分析显示,在有机电解质(dec和dmc)中超声处理之后,s的表面/本体比明显较低。
26.图14:穿过浸泡在dmc中70小时而没有超声处理的具有160nm至3μm范围的不同粒度的单个lgps颗粒的stem eds线扫描,显示硫浓度从表面到本体的变化没有规则模式。
27.图15a
‑
15h:lgps和超lgps颗粒、以及由lgps和超lgps颗粒制成的lib的电化学性能的比较。(a、b)分别为li/lgps/lgps+c/ta单元和li/超lgps/超lgps/ta单元的循环伏安图(cv),所述单元具有锂参比电极,扫描速率为0.1mvs
‑1且扫描范围为0.5v至5v。(c、d)在cv测试之前和之后,小图(a、b)中的lgps单元和超lgps单元的灵敏电化学阻抗谱(eis)。(e、f)在1.0
‑
2.2v的电压范围内以0.5c电流倍率循环的lgps
‑
lib(lto+lgps+c/玻璃纤维隔膜/li)和超lgps
‑
lib(lto+超lgps+c/玻璃纤维隔膜/li)的充电
‑
放电分布。(g、h)lgps lib和超lgps
‑
lib的循环容量曲线。
28.图16a
‑
16b:(a)lgps
‑
asslib(lto+lgps+c作为阴极,lgps作为固态电解质,并且li作为阳极)和(b)超lgps
‑
assl1b(lto+超lgps+c作为阴极,超lgps作为固态电解质,并且li作为阳极)在低电流倍率(0.02c)下的循环性能。
29.图17a
‑
17b:(a)lgps
‑
asslib(lto+lgps+c作为阴极,lgps作为固态电解质,并且li作为阳极)和(b)超lgps
‑
asslib(lto+超lgps+c作为阴极,超lgps作为固态电解质,并且li作为阳极)在中等电流倍率(0.1c)下的循环性能。
30.图18a
‑
18b:(a)lgps
‑
asslib(lto+lgps+c作为阴极,lgps作为固态电解质,并且li作为阳极)和(b)超lgps
‑
asslib(lto+超lgps+c作为阴极,超lgps作为固态电解质,并且li作为阳极)在高电流倍率(0.8c)下的循环性能。
31.图19a
‑
19g:在lgps asslib中循环之后,lto/lgps界面的显微结构和组成(s)tem研究。(a)在1次充电
‑
放电循环之后由lgps asslib制备的fib样品,其中包括阴极层(lto+lgps+c)和se层(lgps)。(b)lto/lgps主界面的tem bf图像,显示具有多个暗颗粒的过渡层。(c)lto颗粒的hrtem图像及其相应的fft图案。(d)lto/lgps主界面的stem df图像显示在过
渡层内的超亮颗粒,指示重元素的累积。(e)穿过主界面进行的stem eels线扫描,指示过渡层内的亮颗粒是富硫的。(f)lto/lgps次界面的stem df图像,其中再次显示较高密度的具有相似形态的亮颗粒。(g)穿过次界面进行的stem eels线扫描,指示亮颗粒是富硫的。
32.图20:lgps
‑
asslib(lto+lgps+c作为阴极,lgps作为固态电解质,并且li作为阳极)的主lto/lgps界面(在阴极和lgps固态电解质层之间的界面)的tem明场图像和stem暗场图像,显示阴极和固态电解质层之间的明显过渡层。
33.图21a
‑
21b:lgps
‑
asslib(lto+lgps+c作为阴极,lgps作为固态电解质,并且li作为阳极)的主lto/lgps界面(在阴极和lgps固态电解质层之间的界面)的(a)stem暗场图像和(b)eels线扫描,显示对于在过渡层内的亮颗粒的内部和外部两者的区域存在li
k
和ge
m4,5
峰。
34.图22a
‑
22b:lgps
‑
asslib(lto+lgps+c作为阴极,lgps作为固态电解质,并且li作为阳极)的主lto/lgps界面(在阴极和lgps固态电解质层之间的界面)的(a)stem暗场图像和(b)eels线扫描,显示s
l1
峰值强度在过渡层内的那些富s明颗粒上更强。
35.图23a
‑
23f:在超lgps asslib中循环之后lto/超lgps界面的显微结构和组成(s)tem研究。(a)lto/超lgps主界面的tem bf图像,显示没有图6b中存在的暗颗粒的光滑界面。(b)对应于图23a中的虚线箭头的stem eels线扫描光谱。(c)lto/超lgps次界面的stem df图像。(d)stem eds线扫描显示硫原子百分比从内部超lgps颗粒到次lto/超lgps界面且最后到lto+c复合区域连续降低。(e)stem eds映射显示图22c中的大颗粒是lgps颗粒。(f)stem eds定量分析显示超lgps颗粒内部的硫原子百分比高达~38%,而次lto/超lgps界面的硫原子百分比低至8%。
36.图24a
‑
24b:另外的(a)stem暗场图像和(b)stem edx线扫描显示,在次lto/超lgps界面处的s浓度比在内部超lgps颗粒区域低得多。
37.图25a
‑
25c:(a)如果评价方案是材料迭代(左列)或元素集迭代(右列),则考虑评价67k种材料的稳定性所需的hull的数目。(b)达到lsps和任意材料a之间的界面稳定性的伪二进制法的图示。表示即使在不存在界面的情况下也存在的材料层面分解能,而g
′
hull
表示由于存在界面而增加的不稳定性。当x=x
m
时,发生最强动力学驱动的反应。d
a
和d
lsps
是在不存在界面的情况下(例如,在x=0,1下)的分解的涂覆材料和lsps。(c)元素分数与增加的化学界面不稳定性的相关性(g
′
hull
(x
m
))。负值是使得浓度增加降低g
′
hull
并改进界面稳定性的那些原子物种。相反地,正值是倾向于增加g
′
hull
和恶化界面稳定性的那些原子物种。由于缺乏大容量数据,仅以少于50种晶体结构存在的元素变灰。
38.图26a
‑
26c:(a
‑
c)分别在0v、2v和4v下元素物种分数与增加的电化学界面稳定性的相关性(g
′
hull
(x
m
))。负值是浓度增加降低g
′
hull
并改进界面稳定性的那些物种。相反地,正值是倾向于增加g
′
hull
和恶化界面稳定性的那些物种。由于缺乏大容量数据,仅以少于50种晶体结构存在的元素变灰。
39.图27a
‑
27d:(a)lsps的hull能量相比相对于锂金属的电压。较深的灰色[中等灰色]阴影突出显示分解为氧化性[还原性]之处。浅灰色阴影表示lsps衰减到不消耗或产生锂(例如,锂中性)的区域。氧化[还原]区域的特征在于hull能量随着电压的增加而增加[减小]。(b)和(c)关于阴离子物种(例如,含氧化合物相对于含硫化合物等、),阳极范围和阴极范围分别的边界电压下的hull能量。中性衰减线之上[之下]的数据点是阳极范围/阴极范
围内的净氧化性[还原性]的。中性衰减线上的那些化合物衰减而不与锂离子储库反应。(d)材料层面电化学分解的平均hull能量相对于电压。
[0040]
图28a
‑
28c:通过阴离子物种分选的化合物的平均lsps界面稳定性的比较。(a)在lsps与所考虑的阴离子类别中的每个之间的化学反应的平均总最大动力驱动能(g
hull
(x
m
))和界面所产生的贡献(g
′
hull
(x
m
))。(b)在给定电压下每种阴离子类别的总电化学不稳定性(g
hull
(x
m
))。(c)在给定电压下界面对每种阴离子类别的电化学不稳定性的平均贡献(g
′
hull
(x
m
))。
[0041]
图29a
‑
29b:通过阴离子物种分选的化合物的功能稳定结果。(a)和(b)确定为功能稳定的每种阴离子类别的总数(线)和百分比(柱)。底部柱表示功能稳定的材料的百分比,且顶部柱表示根据锂化/脱锂的可逆性而潜在功能稳定的材料的百分比。
[0042]
图30a
‑
30f:(a
‑
d)xrd图谱的比较显示在固态电解质材料界面(没有施加电压)处lco、sno2、lto和sio2的结构衰减。在(a)中,
▲
、
●
、
■
、分别代表lco(pdf#44
‑
0145)、lsps(icsd#252037)、sio2(pdf#48
‑
0476)、li3po4(pdf#45
‑
0747)、立方co4s3(pdf#02
‑
1338)、单斜co4s3(pdf#02
‑
1458)。在(b)中,
▲
、
●
、
■
、分别代表sno2(pdf#41
‑
1445)、lsps(icsd#252037)、sio2(pdf#34
‑
1382)、p2s5(pdf#50
‑
0813)和li2s(pdf#23
‑
0369)。在(c)中,
▲
、分别代表lto(pdf#49
‑
0207)、lsps(icsd#252037)和li
1.95
ti
2.05
s4(pdf#40
‑
0878)。在(d)中,
▲
、分别代表sio2(pdf#27
‑
0605)和lsps(icsd#252037)。(a
‑
d)中的阴影区域突出显示在加热至500℃之后发生显著相变之处。界面化学相容性从(a)至(d)降低,分别与lco、sno2、lto和sio2的200mev/原子、97mev/原子、75mev/原子和0mev/原子的预测界面衰减能很好地对应。(e、f)li2s和sio2的cv结果。阴影区域预测该区域中的曲线是否将主要是氧化、还原、中性的。
[0043]
图31a
‑
31e:在室温和500℃下每个单个相:(a)licoo2、(b)lsps、(c)li4ti5o
12
、(d)sno2和(e)sio2的xrd图谱的比较。对于每个相,可以观察到在室温和500℃之间没有显著变化。
[0044]
图32a
‑
32d:在各种温度(室温、300℃、400℃和500℃)下混合物粉末:(a)licoo2+lsps、(b)sno2+lsps、(c)li4ti5o
12
+lsps和(d)sio2+lsps)的xrd图谱的比较。对于licoo2+lsps、sno2+lsps和li4ti5o
12
+lsps,观察到起始反应温度分别为500℃、400℃和500℃。在高达500℃时,观察到sio2+lsps没有发生反应。
[0045]
图33a
‑
33f.(a、b、c)在500℃下热处理36小时之前和之后不同粉末混合物的xrd((a)li+lgps;(b)石墨+lgps;(c)锂化石墨+lgps)。符号和相应的相是:
※
lgps;+li;*石墨;
×
lis2;ges2;
☆
geli5p3。(d)基于lgps的全固态电池中的li/石墨阳极的结构;(e)li/石墨阳极的横截面的sem图像;(f)li和石墨的界面的fib
‑
sem。
[0046]
图34a
‑
34e.(a)li/g
‑
lgps
‑
g/li对称电池和li
‑
lgps
‑
li对称电池之间的循环性能的比较;(b)循环之后对称电池的sem图像。循环300小时之后的li/g
‑
lgps
‑
g/li对称电池(b1,2)和循环10小时之后的li
‑
lgps
‑
li对称电池(b3,4);(c)li/g
‑
lgps
‑
g/li对称电池在不同压力下的倍率性能。(d)在倍率测试之后,在不同压力下li/g
‑
lgps
‑
g/li对称电池的sem图像。(e)li/g
‑
lgps
‑
g/li对称电池的高达10ma/cm2的超高倍率性能。在(e)中施加的压力为250mpa。插图是在
‑
0.3v至0.3v范围内绘制的循环分布,表明在高倍率循环之后没有明显的过电压变化。更多的电压分布放大在补充信息图42中示出。
[0047]
图35a
‑
35d.(a)初始充电/放电曲线的比较,(b)初始库仑效率和(c)在1小时休止之后,在li/g
‑
lgps
‑
lco(linbo3涂覆的)系统中li与石墨的不同容量比之中的开路电压。0、0.5、0.8、1.5、2.5和4的li/g容量比可以分别转化为约0、0.3、0.4、0.8、1.3和2.1的li/g厚度比。在本工作中li/石墨的厚度比默认为1.0
‑
1.3,没有具体的说明。(d)li/g
‑
lgps
‑
lco(linbo3涂覆的)电池的循环性能。
[0048]
图36a
‑
36b.(a)在不同有效模块(k
eff
)下lgps分解的电压分布。(b)对应于不同k
eff
的还原反应途径以及在每个电压范围内不同相平衡中的产物。所有分解产物在此都是每个电压范围内的基态相。
[0049]
图37a
‑
37f.在0.25ma cm
‑2下循环12小时之后,在100mpa下,用聚焦在li/g
‑
lgps
‑
g/li单元中在(a)远离与li/g的界面的中心部分lgps和(b)在li/g和lgps之间的界面上的x射线束对阳极
‑
lgps
‑
阳极对称电池的ge和p的xps测量;(c)在0.25ma cm
‑2下循环10小时之后,在100mpa下,用聚焦在li
‑
lgps
‑
li对称电池中在li和lgps之间的界面上的x射线束对阳极
‑
lgps
‑
阳极对称电池的ge和p的xps测量(失效);(d)在100mpa下,在2ma cm
‑2下倍率测试之后用聚焦在li/g
‑
lgps界面上的x射线束对阳极
‑
lgps
‑
阳极对称电池的ge和p的xps测量;和(e)在250mpa下,在10ma cm
‑2下倍率测试之后用聚焦在li/g
‑
lgps界面上的x射线束对阳极
‑
lgps
‑
阳极对称电池的ge和p的xps测量;(f)在3mpa下,在2ma cm
‑2下倍率测试之后用聚焦在li/g
‑
lgps界面上的x射线束对阳极
‑
lgps
‑
阳极对称电池的ge和p的xps测量。
[0050]
图38.在500℃下加热36小时后,石墨和li与石墨的混合物的xrd。
[0051]
图39a
‑
39c.(a)石墨颗粒的sem图像;在施加高压之后石墨膜的表面(b)和横截面(c)的sem图像。
[0052]
图40.具有相对较小过电压的li/g
‑
lgps
‑
g/li对称电池的循环性能。
[0053]
图41a
‑
44b.li/g阳极在图34(a)中的长期循环之前(a)和之后(b)的sem图像的比较。
[0054]
图42a
‑
42c.(a)li/g
‑
lgps
‑
g/li对称电池的倍率测试。当预循环次数在0.25ma cm
‑2下降低到5次循环时,电池在6ma cm
‑2或7ma cm
‑2下“失效”,然而,当电流密度被设定回0.25ma cm
‑2时,它总是恢复正常而没有显著的过电压增加。(b)放大的图34(e2),以10ma cm
‑2循环的电池,以较小的电压标度(b1)或时间标度(b2)绘制。(c)以不同面积和放大倍数在b中显示的测试之后li/石墨复合材料的sem图像。没有观察到锂枝晶。图42(c2)中是显示这一点的清晰3d结构。
[0055]
图43a
‑
43b.(a)图35d中lco
‑
lgps
‑
li/g电池的循环分布。(b)基于li阳极的循环性能。两个电池都在25℃下在0.1c的电流密度下测试。
[0056]
图44a
‑
44b.来自dft模拟的bader电荷分析。(a)来自分解产物列表的所有p相关化合物中的磷元素;(b)来自分解产物列表的所有ge相关化合物中的ge元素。
[0057]
图45a
‑
45d.(a)在3mpa和100mpa下测试的li/g
‑
lgps
‑
lgps/c电池的cv曲线的比较;(b、c)这两个cv测试之前和之后的阻抗变化的比较;(d)阻抗拟合中使用的模型。r
本体
代表离子扩散电阻,并且r
ct
表示电荷转移电阻。所有eis数据都用z
‑
view拟合。
[0058]
图46a
‑
46g.(a)用1t、3t、6t压制之后的swagelok电池和初始用6t压制的加压单元的cv测试。在阴极中添加了10%的碳。电压范围设定为从开路至9.8v。(b)以放大的电压和电流范围绘制的(a)中的cv扫描。(c)在cv扫描期间对(a)中所示的电池进行的原位阻抗测
试。(d)加压单元在没有电化学过程(黑色)、cv扫描至3.2v、7.5v和9.8v之后的同步辐射xrd。所有cv之后使电压在相同的高截止电压下保持10小时,然后放电回到2.5v。绿色线:在cv扫描至3.2v并保持10小时后,在液态电解质中测试的lgps的同步辐射xrd。(e)在2θ=18.5
°
下不同电池的同步辐射xrd峰,显示在高电压cv扫描并保持之后xrd峰加宽。(f)高电压保持之后lgps的应变相对于尺寸加宽分析。点是在7.5v sxrd测量中不同峰的加宽,相应的xrd峰示于图52中。尺寸和应变加宽的角度依赖性由虚线表示。(g)高电压cv扫描和保持之后s(g1)和p(g2)的xas测量。(g3)在c
‑
方向应变之后p xas峰位移的模拟。
[0059]
图47a
‑
47d.(a)在不同有效模块(k
eff
)下lgps分解能(a1)、基态压力(a2)和基态容量相对于电压。(b)在不同k
eff
下的分解反应途径,以及由不同电压范围内的不同相平衡诱导的产物。(c、d)以下的s(c)和p(d)元素的xps测量:原始lgps(c1,d1),在液态电解质中进行3.2v cv扫描之后的电池(c2,d2),在3.2v cv扫描之后的加压单元(c3,d3)和在9.8v cv扫描后的加压单元(c4,d4)。每次cv扫描之后是在高截止电压下保持10小时。
[0060]
图48a
‑
48e.相对于lto使用(a1)lco、(a2)lnmo和(a3)lcmo作为阴极材料的全固态电池的恒电流充电和放电电压曲线。对于lco、lnmo和lcmo,电池的循环性能分别表示在(b1)、(b2)和(b3)中。在此,lco和lnmo在0.3c下充电和放电,而lcmo在0.3c下充电且在0.1c下放电。所有电池都在室温下,在初始用6t压制的加压单元中测试,并且活性材料用linbo3涂覆,如图54中所示。(c、d)在5次循环之前和之后lco、lnmo、lcmo
‑
lgps的xps测量。(e)对于元素s在5次循环之前(e1)和之后(e2)lco、lnmo、lcmo
‑
lgps的xas测量。
[0061]
图49a
‑
49g.(a
‑
d)在lgps和(a)lno、(b)lco、(c)lcmo、(d)lnmo之间的界面的赝相模拟。图形描绘了界面的反应能相对于所消耗的非lgps相的原子分数。具有最剧烈分解能的原子分数的值定义为x
m
。(e
‑
g)lgps
‑
lno中间相的机械诱导的亚稳图形(由图49a中的分解产生的产物集)。(e)中间相的超出hull的能量(energy over hull)显示出对机械压缩的显著响应。(d)和(e)显示与对于本体相lgps观察到的对压力的类似行为和对压力的容量响应(图47a
‑
47d)。
[0062]
图50a
‑
50c.(a)使用lco和lcmo作为阴极并且石墨涂覆的锂金属作为阳极的全固态电池的恒电流充电和放电分布,截止电压为2.6
‑
4.5v(lco)和2.6
‑
(6
‑
9)v(lcmo)。电池在0.3c下充电且在0.1c下放电。使用(b)在ec/dmc中的1m lipf6和(c)约束的lgps作为电解质的lcmo锂金属电池的循环性能,截止电压为2.5
‑
5.5v,充电倍率为0.3c且放电倍率为0.1c。
[0063]
图51.响应所施加的力的球粒厚度变化。球粒的初始厚度为756μm,球粒的重量为0.14g,球粒的面积为1.266cm2,球粒的经压缩厚度为250μm。计算密度为2.1g/cm3,这与lgps的理论密度2g/cm3接近。
[0064]
图52a
‑
52f.(a)
‑
(f)电池在不同2θ角度下的同步辐射xrd峰,显示在高电压cv扫描和保持之后xrd峰的加宽。加压单元在3.2v cv扫描和保持之后没有显示xrd加宽。
[0065]
图53.(顶部)分解前沿传播的图示。分解相用α...γ标记。可以看出这样的传播需要切向离子传导。(底部)反应坐标的能量图景。最终结果是gibbs能移位δg,基于等式2,其为正的或负的。即使当δg为负的(反应在热力学上是有利的)时,由于切向电流而存在的足够的过电压也可以显著降低前沿的传播速率。
[0066]
图54.linbo3涂覆的lco的stem图像和eds图。
[0067]
图55.使用lgps薄膜作为电解质的lco
‑
lto电池的倍率测试,电池是在0.3c至2.5c
下测试。
[0068]
图56.对于元素p在5次循环之前(表示为p)和之后(表示为5c)的lco、lnmo、lcmo
‑
lgps的xas测量。
[0069]
图57a
‑
57b用swagelok、al加压单元和不锈钢(ss)加压单元测试的使用lgps作为电解质的lco全固态电池的(a)充电分布和(b)放电分布,其中电压截止在3v和4.15v之间。swagelok几乎不施加压力;与不锈钢相比,al单元是软的,并且其施加低约束,而不锈钢在电池测试期间施加最强的恒定约束。
[0070]
图58a
‑
58b.lgps+阴极和lgps+c的cv电流密度的比较。在加压单元中lgps+lco(30+70)(a)和lgps+lcmo(30+70)(b)的cv测量以及在加压单元中lgps+c(90+10)的cv测量。
[0071]
图59a
‑
59d用(a)裸锂金属、(b)石墨和(c)石墨涂覆的li作为阳极组装的lcmo/lgps/li全固态电池。(d)使用不同阳极的lcmo固态电池的循环性能。在第一次循环,所有三个样品都可以充电到约120mah/g,而显然li/石墨在约83mah/g下显示出最高放电容量。可以清楚地看出,li和石墨阳极两者在最初的5次循环内和在20次循环后都有快速衰退的问题,它们的容量都降到20mah/g之下。相比之下,li/石墨阳极的容量得以维持。
具体实施方式
[0072]
本发明提供了可再充电的电池,其包括布置在两个电极之间的、含有碱金属和硫化物的固态电解质(sse)。所述固态电解质可以具有核
‑
壳形态,从而在电压循环条件下赋予增加的稳定性。本发明的这些电池是有利的,因为它们可以是全固态电池,例如,不需要液态电解质,并且可以在电解质降解极少的情况下实现较高的电压。
[0073]
已经表明,其中陶瓷
‑
硫化物固态电解质的核被包裹在刚性无定形壳中的核
‑
壳形态改进了稳定窗口。据信这种稳定背后的机制与陶瓷
‑
硫化物在衰减期间膨胀高达超过20%的趋势有关。应用体积约束机制,这种膨胀被阻止,这又抑制了衰减。我们已经概括了这种理论,并且提供了使用lgps的核
‑
壳形态的合成后产生来显示稳定性改进的实验证据。基于衰减形态,稳定的量值将变化。广义应变模型的平均场解据显示是应变诱导的稳定性的下限。研究的第二衰减形态(成核衰减)据显示提供更大的稳定能力。此外,实验证据提示,衰减实际上是该后一(成核)形态,导致陶瓷
‑
硫化物全电池的显著潜力。
[0074]
支撑核
‑
壳电解质的增强的稳定性和性能的理论的进一步发展已经揭示,应变稳定机制不限于材料层面,而是还可以通过外部应力或体积压缩而应用于电池单元层面。由核
‑
壳结构提供的应变通过局部能垒来稳定固态电解质,这防止发生整体分解。由局部能垒提供的这种稳定效应也可以通过从电池单元施加外部应力或体积压缩来产生,其中对于lgps可以获得高达5.7v的电压稳定窗口,如图1a
‑
1b中所示。在基于这种技术的电池单元设计中,在较高压力和体积压缩下可以预期超过5.7v的较高电压稳定窗口。
[0075]
在固态电池中,当施加的电流密度高于临界值时,形成锂枝晶。在约10mpa的外部压力下,临界电流密度通常报道为1
‑
2ma em
‑2。在本发明中,通过机械压缩改变固态电解质(例如lgps)在阳极界面处的分解途径,并且抑制锂枝晶的生长,导致优异的倍率和循环性能。在电池以高达10ma em
‑2的电流密度循环之后,没有观察到短路或锂枝晶形成。
[0076]
固态电解质
[0077]
本发明的可再充电的电池包含固态电解质材料和掺入固态电解质材料内的碱金
属原子。特定地,用于本发明的电池的固态电解质可以具有核
‑
壳形态,其中核和壳通常具有不同的原子组成。
[0078]
合适的固态电解质材料包括硫化物固态电解质,例如si
x
p
y
s
z
,例如,sip2s
12
,如li
10
sip2s
12
,或β/γ
‑
ps4。其他固态电解质包括但不限于锗固态电解质,例如ge
a
p
b
s
c
,例如gep2s
12
,如li
10
gep2s
12
,锡固态电解质,例如sn
d
p
e
s
f
,例如snp2s
12
,碘固态电解质,例如p2s8i晶体,玻璃电解质,例如碱金属硫化物
‑
p2s5电解质或碱金属硫化物
‑
p2s5‑
碱金属卤化物电解质,或玻璃
‑
陶瓷电解质,例如碱金属p
g
s
h
‑
i
电解质。另一种材料包括li
9.54
si
1.74
p
1.44
s
11.7
cl
0.3
。其他固态电解质材料是本领域已知的。固态电解质材料可以为各种形式,例如粉末、颗粒或固体片材。示例性形式是粉末。
[0079]
可用于本发明电池中使用的固态电解质的碱金属包括li、na、k、rb和cs,例如li。含li固态电解质的实例包括但不限于锂玻璃,例如xli2s
·
(1
‑
x)p2s5,例如2li2s
·
p2s5和xli2s
·
(1
‑
x)p2s5‑
lii,和锂玻璃
‑
陶瓷电解质,例如li7p3s
11
‑
z
。
[0080]
电极材料
[0081]
可以选择电极材料以具有用于离子传输的最佳性质。用于固态电解质电池的电极包含金属,例如过渡金属,例如au,碱金属,例如li,或结晶化合物,例如钛酸锂,如li4ti5o
12
(lto)。阳极也可以包含石墨复合材料,例如锂化石墨。用作固态电解质电池中的电极的其他材料是本领域已知的。电极可以是材料的固体件,或者可以沉积在合适的衬底(例如含氟聚合物或碳)上。例如,在制备用于沉积到衬底上的电极材料的溶液时,已经将液化聚四氟乙烯(ptfe)用作粘合剂。其他粘合剂是本领域已知的。可以使用没有任何添加剂的电极材料。或者,电极材料可以具有添加剂以增强其物理和/或离子传导性质。例如,电极材料可以具有改变暴露于固态电解质(如碳)的表面区域的添加剂。其他添加剂是本领域已知的。
[0082]
4伏licoo2(lco,示于图2a
‑
2b中)和4.8v lini
0.5
mn
1.5
o4(lnmo,示于图3a
‑
3b中)的高电压阴极被证明在本发明的全固态电池中运行良好。更高电压阴极,例如5.0v li2c
o
po4f、5.2v linipo4、5.3v li2ni(po4)f以及6v limnf4和lifef4也可以用作本发明的全固态电池中的电极材料。可以实现超过5.7v(例如高达8v或10v或甚至更高)的电压稳定窗口。另一个阴极是lico
0.5
mn
1.5
o4(lcmo)。示例性阴极材料列于表1中,表1中电极的计算稳定性示于图4中。
[0083]
表1:具有单个材料项目标识符的高电压(大于6v)电极候选物。
[0084]
[0085]
[0086][0087]
电极涂层
[0088]
在一些情况下,电极材料还可以包括在其表面上充当基础电极材料与固态电解质
之间的界面层的涂层。特定地,涂层被配置成改进电极(例如,阴极)与固态电解质之间的界面稳定性,以获得优越的循环性能。例如,用于本发明的电极的涂覆材料包括但不限于石墨、linbo3、alf3、mgf2、al2o3和sio2,特别是linbo3或石墨。
[0089]
基于有效地实现对非常大的数据集的计算搜索的新高通量分析方案,搜索了不同材料的库以找到可以最佳地稳定硫化物固态电解质与典型电极材料之间的界面的那些涂覆材料,其中使用li
10
sip2s
12
作为实例,以预测具有所需化学稳定性和电化学稳定性两者的超过1,000种用于阴极的涂覆材料和超过2,000种用于阳极的涂覆材料。这些通常适用于lgps。表2提供了预测的有效涂覆材料。
[0090]
[0091]
[0092]
[0093]
[0094]
[0095]
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102]
[0103]
[0104]
[0105]
[0106]
[0107]
[0108]
[0109]
[0110]
[0111]
[0112]
[0113]
[0114]
[0115]
[0116]
[0117]
[0118]
[0119]
[0120]
[0121][0122]
外部应力
[0123]
用于增强电解质稳定性的应变稳定机制不限于材料层面,而是也可以通过外部应
力或体积压缩而应用于电池单元层面。在某些实施方案中,外部应力是例如通过机械压制递送的、施加到可再充电的电池的全部或一部分(例如,固态电解质)的体积约束。外部应力可以通过例如由金属制成的壳体施加。在一些情况下,体积约束可以是约70mpa至约1,000mpa,例如约70mpa至约150mpa、约100mpa至约300mpa、约200mpa至约400mpa、约300mpa至约500mpa、约400mpa至约600mpa、约500mpa至约700mpa、约600mpa至约800mpa、约700mpa至约900mpa或约800mpa至约1,000mpa,例如约70mpa、约75mpa、约80mpa、约85mpa、约90mpa、约95mpa、约100mpa、约150mpa、约200mpa、约250mpa、约300mpa、约350mpa、约400mpa、约450mpa、约500mpa、约550mpa、约600mpa、约650mpa、约700mpa、约750mpa、约800mpa、约850mpa、约900mpa、约950mpa或约1,000mpa。在本发明中,“约”是指
±
10%。
[0124]
固态电解质也可以在包含在电池中之前被压缩。例如,固态电解质可以用约70mpa至约1,000mpa,例如约70mpa至约150mpa、约100mpa至约300mpa、约200mpa至约400mpa、约300mpa至约500mpa、约400mpa至约600mpa、约500mpa至约700mpa、约600mpa至约800mpa、约700mpa至约900mpa或约800mpa至约1,000mpa,例如约70mpa、约75mpa、约80mpa、约85mpa、约90mpa、约95mpa、约100mpa、约150mpa、约200mpa、约250mpa、约300mpa、约350mpa、约400mpa、约450mpa、约500mpa、约550mpa、约600mpa、约650mpa、约700mpa、约750mpa、约800mpa、约850mpa、约900mpa、约950mpa或约1,000mpa的力压缩。一旦被压制,固态电解质就可以被用于电池中。这样的电池也可以经受外部应力以对固态电解质(例如,在显微结构层面上)施加机械压缩(mechanical constriction),即提供等容约束。固态电解质上的机械压缩可以是1gpa至100gpa,例如5gpa至50gpa,如约15gpa。维持机械压缩所需的外部应力可以是约1mpa至约1,000mpa,例如约1mpa至约50mpa、约1mpa至约250mpa、约3mpa至约30mpa、约30mpa至约50mpa、约70mpa至约150mpa、约100mpa至约300mpa、约200mpa至约400mpa、约300mpa至约500mpa、约400mpa至约600mpa、约500mpa至约700mpa、约600mpa至约800mpa、约700mpa至约900mpa或约800mpa至约1,000mpa,例如约70mpa、约75mpa、约80mpa、约85mpa、约90mpa、约95mpa、约100mpa、约150mpa、约200mpa、约250mpa、约300mpa、约350mpa、约400mpa、约450mpa、约500mpa、约550mpa、约600mpa、约650mpa、约700mpa、约750mpa、约800mpa、约850mpa、约900mpa、约950mpa或约1,000mpa。所采用的外部应力可以根据电池的电压而改变。例如,在6v下工作的电池可以采用约3mpa至约30mpa的外部应力,并且在10v下工作的电池可以采用约200mpa的外部应力。本发明还提供了在将固态电解质包括在电池中之前使用固态电解质的压缩,例如随后施加外部应力,来制造电池的方法。
[0125]
方法
[0126]
本发明的电池可以被充电和放电期望的循环次数,例如1至10,000次或更多次。例如,电池可以循环10至750次或至少50、100、200、300、400、500、600、700、800、900、1,000、1,500、2,000、3,000、4,000或5,000次。在实施方案中,电池的电压范围为约1v至约20v,例如约1
‑
10v、约5
‑
10v或约5
‑
8v。本发明的电池还可以在例如1ma cm
‑2至20ma cm
‑2,例如约1
‑
10ma cm
‑2、约3
‑
10macm
‑2或约5
‑
10ma cm
‑2的任何合适的电流密度下循环。
[0127]
实施例
[0128]
实施例1
[0129]
在solartron电化学恒电位仪(1470e)上,使用锂(由li2hpo4涂覆)作为参比电极,在开路电压(ocv)至6v之间以0.1mvs
‑1的扫描速率在不同压力下测量li/lgps/lgps+c的循
环伏安图(cv)。还组装了使用lgps/c薄膜作为阴极、锂作为阳极和在ec/dmc中的1m lipf6作为电解质的液态电池以供比较。在固态cv测试和液态cv测试两者中lgps与c的比率均为10∶1。
[0130]
通过将lto/lco/lnmo、lgps、聚四氟乙烯(ptfe)和炭黑以不同的重量比混合来制备用于全固态电池的阴极和阳极薄膜。对于lto、lco和lnmo薄膜电极,活性材料/lgps/c的比率分别为30/60/10、70/27/3、70/30/0。然后将该粉末混合物在研钵中手工研磨30分钟,并且在填充氩气的手套箱中在添加3%ptfe的情况下滚压成薄膜。通过将lgps和ptfe以97∶3的重量比混合,然后在研钵中手工研磨混合粉末30分钟且最终在填充氩气的手套箱中将其滚压成薄膜来制备用于全固态锂离子电池的固态电解质。为了组装全固态锂离子电池单元,将制备的复合阴极(lco或lnmo)薄膜、lgps薄膜(<100μm)和阳极(lto)薄膜分别用作阴极、固态电解质和阳极。将阴极、电解质和阳极的三个薄膜在420mpa下冷压在一起,并且在电池循环测试期间通过使用加压单元将压力保持在210mpa。在室温下使用arbinbt2000工作站(arbin instruments,tx,usa)测试充电和放电行为。基于lto的量计算比容量。
[0131]
实施例2
‑
应变稳定的lgps核
‑
壳电解质电池
[0132]
理论
‑
物理图景
[0133]
图4a中描绘了应变可以扩展lgps稳定窗口的机制。考虑在标准温度和压力下,lgps分解为某一任意分解产物集,表示为“d”(lgps
→
d)。作为已经分解的lgps的分数(x
d
)的函数的系统的gibbs能由图4a中的橙色虚线给出,并且在等式1中解析地给出。
[0134]
g0(x
d
)=(1
‑
x
d
)g
lgps
+x
d
g
d
ꢀꢀꢀ
(1)
[0135]
最低gibbs能态为x
d
=1(全部分解)且初始态为x
d
=0(原始lgps)。因此,反应能为δg0=g0(1)
‑
g0(0)=g
d
‑
g
lgps
。这种系统固有地不稳定。也就是说,对于所有的x
d
值,均为负。因此,对于x
d
的任何初始值,系统将移动以通过增加x
d
而减小g0,最终在最终状态x
d
=1下结束。
[0136]
接下来,考虑应用约束lgps颗粒的机械系统。考虑到lgps在衰减期间趋于膨胀,任何机械约束都将需要分解在周围邻域中诱导应变。这样的约束系统可以是材料层面(即,核
‑
壳显微结构)或系统层面(即,加压的电池单元)或两者的组合。最终,这种机械系统在破裂之前仅可以诱导有限的应变。使系统破裂所需的能量表示为g
破裂
。
[0137]
在约束机制破裂前,lgps的任何分解必定导致应变能增加。图5a
‑
5b中的绿色线关于无约束的gibbs(g0)和约束诱导的应变(g
应变
)绘制受约束的gibbs能(g
′
)。突出显示的曲线指示lgps的分解途径。
[0138]
1.颗粒作为原始lgps(x
d
=0)开始,具有未破裂的约束机制
[0139]
2.随着颗粒开始分解(x
d
:0
→
δx
d
),约束机制需要g
应变
的增加。假定应变gibbs是x
d
的函数,其随着x
d
趋向零而趋向零
[0140]
3.一旦应变的系统的gibbs能(g
′
(x
d
))超过破裂的系统的gibbs能(g0(x
d
)+g
破裂
),约束机制就将失效。这在破裂点x
d
=x
f
下发生
[0141]
4.一旦x
d
>x
f
,系统就将继续完全分解,
[0142]
如果约束诱导的应变gibbs(g
应变
)足够陡,则总gibbs在x
d
<x
f
下的斜率将是正的(如图5a中所绘)。在这种情况下,lgps将在原始状态(x
d
=0)周围是亚稳的。这项工作集中
在约束系统的量化,使得在x
d
≈0下,允许亚稳的陶瓷硫化物电解质。
[0143]
两个功微分
[0144]
作为x
d
的函数的g
应变
的存在源于lgps在分解时膨胀的性质。取决于如由施加的电压确定的分解产物集,这种体积膨胀可以超过20
‑
50%。因此,lgps分解的过程是可以包括显著的“无应力”应变的过程,即,应变是分解的结果而不是施加的应力的结果。对这样的衰减途径的适当热力学分析需要仔细考虑对于其他系统而言合理忽略的多个功微分。
[0145]
图5b示意性地表示两个经常使用的功源,即“类流体”形式和“类固体”形式。在类流体系统中,等压条件下的功变化与系统体积的变化成比例δw=
‑
pδv。对于类固体系统,功关于参考/未变形状态定义,并且具有微分形式δw=v
ref
σ
ij
δe
ij
,其中v
ref
是未变形的体积,∈是相对于未变形状态的应变张量,并且σ是对应于∈的应力张量。
[0146]
显示这两个微分功表达的等同性的一般方法如下。通过使用如等式2中定义的偏张量,将类固体应力和应变张量分成压缩项和变形项。压力关于应力矩阵和体积应变∈≡(v
‑
v
ref
)/v
ref
而概括。
[0147][0148]
使用这些定义,可以将类固体功分成一个仅包括压缩的项和一个仅包括变形的项。
[0149][0150]
在流体极限中,其中不存在形状变化,假设δv
ref
=0,等式3简化为δw=
‑
v
ref
pδ∈=pδv,恢复了类流体功微分。在大多数机械系统中,这种假设是有效的,因为未变形的参考体积不改变。然而,由于未变形的体积关于x
d
而变化,并且因此δv
ref
≠0,所以描述lgps分解时失效。
[0151]
v
ref
(x
d
)=(1
‑
x
d
)v
lgps
+x
d
v
d
ꢀꢀꢀ
(4)
[0152]
相反,lgps分解的适当热力学分析需要考虑两个功项。流体项
‑
pδv
ref
指示在存在应力张量σ的情况下压缩参考体积(即,改变x
d
)所需的功,并且固体项表示使新的参考状态变形所需的功v
ref
σ
ij
δ∈
ij
。考虑到这一点,全能量微分由等式5给出。
[0153]
δe=tδs+μ
α
n
α
‑
pδv
ref
+v
ref
σ
ij
δ∈
ij
ꢀꢀꢀ
(5)
[0154]
转换成gibbs能g=e
‑
ts+pv
ref
‑
v
ref
σ
ij
∈
ij
=μ
α
n
α
,产生微分形式:
[0155]
δg=
‑
sdt+μ
α
δn
α
+vδp
‑
v
ref
∈
ij
δσ
ij
ꢀꢀꢀ
(6)
[0156]
注意,只要v
ref
恒定,则在固体力学中经常使用的转换g=e
‑
ts
‑
v
ref
σ
ij
∈
ij
=μ
α
n
α
‑
pv
ref
就足够,并且因此
‑
pv
ref
可以被设定为零点。
[0157]
在恒定温度下,等式6关于化学项(δg0=μ
α
δn
α
)和应变项给出图5a
‑
5b的g
′
(x
d
)的微分形式。
[0158][0159]
在以下讨论中,对于作为x
d
的函数的g
应变
,g
应变
提供可以使lgps稳定的值的范围,我
们考虑两种限制情况。第一种情况是lgps颗粒流体静力学分解并且是平均场近似。假设分解的lgps的分数在整个颗粒中是均匀的(对于所有)。第二种限制情况是球形对称成核,其中lgps在半径r
i
的球形区域内完全分解(),而在这一区域之外为原始lgps如下所示,流体静力学情况产生g
应变
的下限,而成核模型显示这个值在实践中可以如何高得多。
[0160]
流体静力学极限/平均场理论
[0161]
lgps颗粒的分区所经历的局部应力直接随分解分布以及颗粒的机械性质和机械约束系统(如果应用的话)而变。在流体静力学近似中,局部应力被说成是压缩性的并且在颗粒内的任何地方都相等在平均场近似中,对于分解分数也是如此考虑到和之间的一一对应关系,则这两个近似是等同的。
[0162]
我们将焦点限制到x
d
→
0的极限,以关于原始状态评价lgps的亚稳性。如果则已知颗粒是至少亚稳的,其中总稳定性由g
破裂
的量值确定。在这个极限内,压力和分解分数之间的关系在参考文献
22
中显示为p(x
d
)=x
d
k
eff
∈
rxn
。其中k
eff
是系统的有效本体模量,考虑了材料的压缩率和施加的机械约束两者。k
eff
指示将需要多大的压力来充分压缩系统以允许lgps的体积膨胀(∈
rxn
),所述体积膨胀伴随着分解。如等式8中所示,假设没有偏差应变(对于流体模型是合理的),可以自此求解出微分应变gibbs。
[0163][0164][0165]
参考体积是无约束系统中的体积,v
ref
=(1
‑
x
d
)v
lgps
+x
d
v
d
。组合等式7和等式9与亚稳条件发现只要满足等式10,类流体lgps就将稳定。
[0166][0167]
在图5中对于核
‑
壳压缩机制的情况求解等式9,其中核由lgps或氧掺杂的lgpso(li
10
gep2s
11.5
o
0.5
)组成且壳为任意刚性材料。有效本体模量由k
eff
=(β
lgps
+β
壳
)
‑1给出,其中β
lgps
为lgps材料的压缩率,并且是表示壳约束颗粒的能力的参数
22
。
[0168]
球形成核极限
[0169]
最大局部(即,最高局部压力)分解机制是如图6中所示的球形成核的机制。在这种模型中,外径r
o
的lgps颗粒在其中心处经历分解。分解的区域对应于初始在半径r
i
内的材料。由于材料已经分解成由给出的更大体积,所以新的参考状态具有比原始状态高的体积。分解分数在颗粒中不再是如在流体静力学情况下的常数。相反,对于初始(分解前)在区域r<r
i
内的所有材料,且对于初始在该区域之外r>r
i
的所有材料,
[0170]
为了将半径r
d
的经分解参考状态适配到半径ri的空隙中,分解的球体和剩余lgps两者都必须如图7a.iii和7a.iv中所示变成应变的。因此,关于分解分数x
d
求解应力成为压缩固态球体的厚壁球形压力容器的问题。压力容器具有由r
i
和r
o
给出的参考状态内径和外
径,并且球形颗粒具有平衡半径r
d
=(1+∈
rxn
)
1/3
r
i
。
[0171]
关于分解材料和原始材料的位移向量和以及径向应力分量和边界条件是:
[0172]
1.分解产物和原始产物之间的连续性:r
d
+u
d
(r
d
)=r
i
+u
p
(r
i
)。其中已经去掉了向量符号以反映系统的径向对称性。
[0173]
2.在分解产物和原始产物之间的界面处那些材料的应力的径向分量之间的连续性:
[0174]
对于各向同性材料中的球形对称应力,已知位移向量具有如下形式u(r)=ar+br
‑2,其中由于位移仅是距中心的距离的函数,因此已经去除了向量符号。条件下压缩球体的应变gibbs给出没有偏分量的压缩项同样,在衰减开始时的中空加压球体具有压缩分量和偏分量两者,它们组合成其中s
p
为原始材料的剪切模量。组合这些项产生等式8的成核当量。
[0175][0176]
图7b显示对于原始材料和分解材料具有相同的弹性模量(e
p
=e
d
)和泊松比(v
p
=v
d
)的情况求解的等式11。灰色线和紫色线反映了流体静力学模型的无壳极限和完美壳极限,而蓝色线和红色线表示典型泊松值的等式10。可以看出,通常,由于涉及更高的压力,成核模型提供比流体静力学模型更陡的应变gibbs。直观地,较小的泊松比(更难压缩)改进成核极限的稳定性。
[0177]
钝化层理论
[0178]
电极电位在电解质稳定窗口之外时,液态或固态的电解质可能与电极反应。为了解决这个问题,建议选择电解质,使得它们形成在电极电位下至少动力学稳定的钝化固态电解质界面(sei)。许多关于改进硫化物电解质的主题的工作已经推测,通过在硫化物电解质的表面上形成电子绝缘层,可以形成这种钝化层。在这一部分中,我们讨论了这种钝化层的作用,并且提供了我们认为电子绝缘表面层改进稳定性的机制的定量分析。
[0179]
在图8a中,对于最基本的电池半电池模型给出了热力学平衡状态。阴极通过电绝缘和离子传导的材料(σ=0,κ≠0,其中σ,κ是电子传导率和离子传导率)与锂金属隔开,并且相对于锂金属向阴极施加电压φ。锂金属的电压被定义为零点。关于电子数(n)、锂离子数(n)、费米能级(ε
f
)和锂离子化学势(μ
li
+),微分gibbs能可以写为等式12(上标a、c区分阳极和阴极)。
[0180][0181]
应用守恒δn
a
=
‑
δn
c
,δn
a
=
‑
δn
c
给出了熟知的平衡条件:
[0182][0183]
或者,换句话说,电子和锂离子两者的电化学势(η=μ+zeφ)在单元内的任何地方必须是恒定的。结果,锂金属电位(μ
li
=η
li
++η
e
‑
)在整个单元中保持恒定。图7a中见到的能带图示出每个物种的化学势以及电压如何在整个单元中变化,而电化学势保持恒定。
[0184]
图8b描绘了在固态电解质阴极的情况下的预期平衡状态,其中阴极材料嵌入固态电解质的基质中。在这种情况下,阴极材料相对于电解质的较低(即更负)化学势引起电荷分离,这产生界面电压χ
i
。类似于遵循等式12的程序,它可以表明平衡点现在包括阳极(a)、阴极(c)和固态电解质(se):
[0185][0186]
与等式13类似,等式14得出锂金属电位在整个单元中保持恒定的条件。
[0187]
硫化物电解质的钝化层稳定的推测机制描绘在图8c中。在这种情况下,固态电解质被电子绝缘材料涂覆。由于外部电路不直接接触固态电解质,并且不存在电子传导途径,因此固态电解质内的电子数目是固定的。因此,费米能量不能通过电子流平衡。据推测,这种效应可以用于允许固态电解质内的锂金属电位相对于电极的偏差,导致更宽的工作电压窗口。图8c的能带图示出由于缺乏电子传导,电子电化学势如何能够在固态电解质中经历局部最大值(或最小值)。这种局部最大值(或最小值)被转入到锂金属电位。
[0188]
作者相信,虽然电子绝缘钝化层是关键的设计参数,但是上述理论缺少有效电子传导的关键作用,所述有效电子传导由于在锂离子迁移出绝缘区域而留下相应电子时产生的“锂空穴”而发生。这种系统的微分gibbs能通过将固态电解质项加到等式12(由上标se表示)来表示。
[0189][0190]
电子和锂守恒约束现在是:
[0191]
1.δn
se
=
‑
δn
se
:从se中去除锂离子的效应是将相应电子置于剩余材料的费米能级。
[0192]
2.δn
a
=
‑
δn
c
+δn
se
:在阳极获得锂离子,而不是相应的电子,这降低处于费米能级的电子的数目。
[0193]
3.δn
a
=
‑
δn
c
‑
δn
se
:总锂守恒。
[0194]
约束1和2表示在绝缘颗粒的情况下电子密度和锂密度的拘束(tethering)。与由等式12支配的系统不同,固态电解质的费米能级不是由外部电压固定的。结果是,通过提取锂离子来减少固态电解质内原子的数目,并且因此增加绝缘区域内每原子的电子数、每原子的电子数和费米能级增加。实际上,这表示电子通过锂空穴的传导。对给出上述约束的平衡点求解等式15导致在阳极/阴极之间的等式14的那些以及在阳极和固态电解质之间的以下关系。
[0195][0196]
se内经历的总电压可以表示为其中是在不存在从se中的锂提取的情况下的电压(如图8c中所绘的原始电压),并且v
s
是由锂提取的电荷分离产生的电压。换句话说,系统以电压下电荷中性的固态电解质开始。然而,通常不满足等式16。电荷分离发生,降低了固态电解质相对于阳极的电压。关于几何确定的电容c,所述电荷分离电压是v
s
=c
‑1en
se
。这种效应在图8d中示出。在se区内的电荷分离前,由蓝色实线给出电压和化学势。随着通过阳极从se中提取锂离子,se中的电压从降至
[0197]
在电子绝缘区域内的这种电压弛豫的最终结果描绘在图8e中。由于经由锂空穴传导的有效电子传输,一旦施加的电压超过固态电解质的固有稳定性,带负电荷的锂金属就可以在颗粒内局部形成。负电荷是由于锂离子离开绝缘区域以平衡锂金属电位。因此,预期局部(即,在绝缘区域内)锂金属与剩余固态电解质具有界面电压χ
i
。该电压必须等于在阳极锂和固态电解质之间的电压χ
i
=φ
se
。简而言之,从热力学角度来看,相对于锂金属阳极向电子绝缘的固态电解质颗粒施加电压φ
se
等同于施加与固态电解质直接接触的带电荷锂金属。
[0198]
本质上,这对固态电解质稳定性没有影响。然而,在非常低的电容的极限内,如所预期的,仅一小部分的锂离子将需要迁移到阳极,因为因此,电子绝缘壳局部地捕获大部分的锂离子,这维持机械稳定所需的高反应应变。
[0199]
结果和讨论
[0200]
电化学稳定性
[0201]
通过比较在lgps与具有提供压缩机制的添加的核
‑
壳形态的相同lgps之间的衰减度量,研宄机械压缩对lgps稳定性的影响。为了使化学变化最小化,使用合成后超声处理产生压缩性核
‑
壳形态。这种核
‑
壳lgps(下文称为“超lgps”)通过高频超声处理实现,所述高频超声处理引起lgps的外层向无定形材料转化。lgps颗粒在超声处理之前(图9a)和在超声处理之后(图9c)的明场透射电子显微镜(tem)图像显示无定形层的明显形成。统计分析的能量色散x射线光谱(eds)(图9b和9c)显示这种无定形壳是略微缺硫的,而lgps和超lgps的本体区域维持几乎相同的元素分布。对单个[超]lgps颗粒进行的eds线扫描(图10
‑
12)证实,几乎每个超lgps颗粒都存在缺硫表面层,而对于lgps颗粒没有观察到这种现象。注意,对于lgps在两种测试溶剂碳酸二甲酯(dmc)和碳酸二乙酯(dec)中的超声处理,情况都是如此(图11
‑
13)。将lgps简单地浸泡在dmc中而不进行超声处理没有明显的效果(图14)。这种合成后核
‑
壳形成的方法使lgps的本体结构变化最小化,从而允许我们评价体积压缩对稳定性的影响而没有组成变化。
[0202]
分别使用li/lgps/lgps+c/ta单元(图15a)和li/超lgps/ta单元(图15b)的循环伏安法(cv)测量来评价未压缩的lgps和压缩的超lgps的电化学稳定性,其中使用锂参比电极,扫描速率为0.1mvs
‑1且扫描范围为0.5
‑
5v。在此引入碳以测量电解质的固有电化学稳定窗口而没有动力学妥协。
12
对于lgps,在充电期间观察到在2
‑
4v和3.7v下的氧化峰,并且在放电期间观察到在1.6v之下的多个峰。这些氧化还原峰可以归因于lgps中li
‑
s和ge
‑
s组分
的固
‑
固相转变
24
,证实lgps不稳定且在循环期间发生严重分解。
[0203]
相反,超lgps的分解极大地被抑制,显现为在充电期间在较高电压(3v)下仅有一个较小的氧化峰,而在放电期间几乎没有还原峰(图15b)。事实上,在cv测试之前和之后的灵敏电化学阻抗谱(eis)(图15c、15d)也证实了超lgps的较高稳定性。eis显示类电池行为的典型奈奎斯特图形(nyquist plot),在中等频率下具有电荷转移半圆,并且在低频率下具有扩散线。结果表明,在cv测试的3次循环后,lgps复合材料的总阻抗从300q增加到620q(增加107%)(图15c),而超lgps复合材料的总阻抗仅增加32%(从250ω增加到330q,图15d)。循环之后阻抗的较小增加指示超lgps更稳定,因此较少的固相和晶界由于分解而产生。
[0204]
发现当在全固态半电池电池组中实施时,超lgps优于lgps的这些稳定性优势甚至更突出。测量与碳混合的li4t5o
12
(lto)的循环性能,其中使用超lgps或lgps作为阴极,超lgps或lgps作为隔膜且锂金属作为阳极。在低电流倍率(0.02c)、中等电流倍率(0.1c)和高电流倍率(0.8c)下获得每种配置的循环性能。在图16a
‑
18b中描绘的结果表明基于超lgps的半电池的循环稳定性明显超过基于lgps的半电池。
[0205]
为了分离出lto阴极复合材料中lgps的分解,固态电解质层被玻璃纤维隔膜代替。图15e显示在0.5c下在1.0
‑
2.2v的电压范围内循环的lgps(lto+lgps+c/玻璃纤维隔膜/li)的充电
‑
放电分布。在70次循环中出现在1.55v下的平坦电压平台,这可以归因于钛的氧化还原。然而,平台长度从循环1至循环70降低了几乎85.7%,指示阴极的大衰减。另一方面,超lgps(lto+超lgps+c/玻璃纤维隔膜/li)(图15f)显示相同的平坦电压平台在70次循环之后保持几乎不变。循环容量曲线(图15g和15h)进一步证实阴极稳定性的这种增加。对于lgps,在70次循环后,充电和放电比容量分别从~159mah/g降低至~27mah/g和从~170mah/g降低至~28mah/g。然而,超lgps展示比其lgps对应物好得多的循环稳定性。在70次循环之后,放电容量仍高达160mah/g,仅有粗略的5%的容量损失。
[0206]
在这些结果的每一个中,具有核
‑
壳形态的那些超lgps颗粒都胜过lgps对应物的稳定性。如参考文献
22
中所讨论,提出了核
‑
壳设计,以通过由壳对核的体积约束来稳定陶瓷
‑
硫化物固态电解质。本实验电化学稳定性数据与这一理论一致。如在超lgps的情况下所见,预期缺硫壳降低系统的有效压缩率,并且因此增加体积约束
22
。在相对于锂的1
‑
2.2v的电压范围中固态半电池(固态阴极+玻璃纤维/液态电解质+锂金属阳极)性能展示,在lgps氧化和还原两种情况下,超lgps实际上都具有比lgps改进的稳定性。另外,超lgps的库仑效率也高于lgps,指示系统中的电荷转移效率改进,并且较少的电荷参与不需要的副反应。
[0207]
分解机制
[0208]
为了更好地理解lgps分解的机制,进行tem分析以研究循环之后lto/[超]lgps界面的显微结构。在相对于锂金属阳极的1次充电
‑
放电循环之后,制备fib样品(图19a),其中包括复合阴极(lto+lgps+c)和隔离层(lgps)。在fib样品制备期间将铂层沉积到阴极层上以保护其免受离子束研磨。在阴极/隔膜界面(下文称为“lto/lgps主界面”)存在具有多个小的暗颗粒的过渡层,如tem明场(bf)图像(图19b、图20)和stem暗场(df)图像(图19d、图20)中所显现。stem df图像的过渡层内的颗粒显示明反差,指示重元素的累积。为了理解所述过渡层的化学组成,进行stem eels(电子能量损失光谱)线扫描。eels光谱显示li
k
、ge
m4,5
(图21a
‑
21b)、ge
m2,3
和p
l2,3
(图15e)峰存在于整个过渡层中,但硫峰(s
l2,3
、s
l1
)仅在亮颗粒内
部出现,并且在亮颗粒外部的区域不存在(图15e中的eels光谱12
‑
14)。这一观察结果指示,过渡层内的亮颗粒是富硫的,这不仅得到stem图像(硫是li、ge、p和s中最重的元素)和eels线扫描观察(图19e、图21a、图21b、图22a和图22b)中明反差的支持,而且得到了报道lgps的分解产物将是包括s、lis、p2s5和ges2的富硫相的先前研究
12
的确认。
[0209]
由于复合阴极层由lto、lgps和c组成,因此将存在在阴极层内普遍存在的次要lto/lgps界面(下文称为“lto/lgps次界面”)。图19f展示lto/lgps次界面的典型stem df图像,其中再次出现具有相似形态的亮颗粒。这种亮颗粒的密度由于阴极层内较高的碳浓度而高得多,并且因此促进lgps分解。相应的stem eels线扫描光谱(图19g)显示强s
l2,3
峰存在于界面区域,再次确认了亮颗粒是富硫的。因此,在1次充电
‑
放电循环之后,富硫颗粒存在于lgps半电池中的主lto/lgps界面和次lto/lgps界面两者处。
[0210]
作为比较,图23a
‑
23f显示超lgps半电池的显微结构和组成(s)tem研究。在1次充电
‑
放电循环之后的主lto/超lgps界面通过tem bf图像(图23a)表征。在超lgps隔离层和复合阴极层之间观察到光滑的界面(图23b)。主lto/超lgps界面干净且均匀,显示没有过渡层或暗颗粒。还通过stem df图像、eds线扫描和eds映射研究次lto/超lgps界面以进行比较(图23c
‑
23e)。结果显示,随着stem eds线扫描从内部超lgps颗粒进行到次lto/超lgps界面,最后进入lto+c复合区域(图23d和图24a、图24b),硫的原子百分比不断降低。换句话说,超lgps颗粒在循环之后仍维持缺硫壳特性,并且在lto/超lgps次界面处不形成富硫过渡层。stem eds定量分析(图23f)显示,超lgps颗粒内部的硫原子百分比高达~38%,而次lto/超lgps界面的硫原子百分比低至8%。
[0211]
这些结果提示,与流体静力学极限相比,成核极限更真实地代表了真正的衰变过程。lgps中形成的富硫颗粒具有r
i
≈20nm数量级的长度标度。在超lgps中,壳厚度也是粗略的l≈20nm。因此,如果我们考虑在超lgps中的核
‑
壳边界附近形成这种硫颗粒,则从富硫颗粒的中心到壳外部的最小距离是r
o
=r
i
+l≈40nm。在这种情况下,其满足应用成核模型所需的条件r
i
<<r
o
。总之,我们知道lgps通过导致富硫颗粒在表面上成核的机制而衰减。我们还知道,应用具有某一厚度的壳层使得l≈r
i
抑制这种衰减。这些结果提示,关于朝向在核
‑
壳界面正下方具有成核衰减的状态的衰减,原始的核
‑
壳状态至少是亚稳的。
[0212]
结论
[0213]
总之,我们已经开发了广义应变模型,以显示考虑到lgps在衰减时膨胀的性质,机械压缩如何能够导致在显著扩展的电压范围内的亚稳性。压缩扩展电压窗口的精确水平取决于衰减的形态。我们对衰减形态的两个极限:最小局限情况和最大局限情况进行了理论分析。最小局限情况由平均场理论组成,其中颗粒的每个部分同时衰减,而最大局限情况由成核衰减组成。已经证明,尽管最大局限情况最好,但两种情况都具有大大扩展稳定窗口的潜力。我们还开发了电绝缘钝化层在这种应变稳定的系统中的作用的理论。这种模型提示,这种钝化层通过保持锂离子局限于颗粒内、使反应应变最大化而有助于稳定性。
[0214]
增加压缩性壳之前和之后lgps的稳定性能的实验结果支持这种理论。在通过超声处理形成壳之后,lgps展示性能显著改进的循环伏安法、固态电池循环和固态半电池循环。因为壳是在合成后方法中施加的,所以使核
‑
壳和纯lgps样品之间的化学差异保持最小,否则这种化学差异可能影响稳定性。据信核
‑
壳是机械约束的lgps的实例,因为在任何分解期间,lgps核都将试图膨胀,而壳将保持固定。换句话说,壳对核提供准等容约束,这取决于壳
的双轴模量和颗粒几何形状。
[0215]
对在lgps颗粒中而不是在超lgps颗粒中见到的衰减形态的分析提示,成核衰减极限更准确地反映真实的热力学。发现在lgps中,在循环之后成核的富硫衰减中心嵌入lgps颗粒的表面。另外,在经循环的超lgps中没有见到这些成核的衰减中心。超lgps维持与lgps中的衰减位点相当的壳厚度(大约20nm),预测这对于由成核模型提供的高水平稳定是足够的。这些结果,与超lgps的改进的稳定性组合,指示不仅发生应变稳定,而且发生应变稳定的量值由最大局限衰减机制主导。这是一种有前景的结果,因为已经显示这种成核衰减提供较大的g
应变
值,为在比迄今为止所报道的电压高得多的电压下工作的固态电池打开了大门。
[0216]
方法
[0217]
样品制备
[0218]
lgps粉末购自mse supplies公司。通过将lgps粉末浸泡到有机电解质(如碳酸二甲酯(dmc)和碳酸二乙酯(dec))中,然后在来自qsonica公司的q125超声发生器(一种基于微处理器的可编程超声处理器)中超声处理70小时,合成超lgps。
[0219]
电化学
[0220]
在solartron电化学恒电位仪(1470e)上,使用锂作为参比电极,在0.5v和5v之间以0.1mvs
‑1的扫描速率测量li/lgps/lgps+c/ta单元和li/超lgps/超lgps/ta单元的循环伏安图(cv)。在cv测试之前和cv测试之后,通过在1mhz至0.1hz频率范围内施加50mv振幅ac电位,在室温下测量li/lgps/lgps+c/ta单元和li/超lgps/超lgps/ta单元的电化学阻抗谱。通过将lto、(超)lgps、聚偏二氟乙烯(pvdf)和炭黑以30∶60∶5∶5的重量比混合来制备所用的复合阴极。然后将这种粉末混合物在研钵中手工研磨30分钟,并且在填充氩气的手套箱中滚压成薄膜。通过将(超)lgps和pvdf以95∶5的重量比混合,然后在研钵中手工研磨混合粉末30分钟且最终在填充氩气的手套箱内将其滚压成薄膜来制备se。为了组装固态单元,将制备的复合阴极薄膜、(超)lgps薄膜和li金属箔分别用作阴极、固态电解质和对电极。在组装成电池之前,将复合阴极和(超)lgps的薄膜冷压在一起。将一片玻璃纤维隔膜插入(超)lgps薄膜和li金属箔之间,以避免在这两个相之间的界面反应。仅将1滴在碳酸亚乙酯(ec)和碳酸二甲酯(dmc)溶液(1:1)中的1m lipf6小心地施加到玻璃纤维上,以允许锂离子传导通过隔膜。将swagelok型单元在填充氩气的手套箱内组装。(超)lgps电池的组装过程与(超)lgps固态电池的组装过程相同,不同之处在于(超)lgps se层被去除。在室温下使用arbinbt2000工作站(arbin instruments,tx,usa)测试充电/放电行为。比容量基于阴极膜中lto的量(30重量%)计算。
[0221]
表征
[0222]
对于fib样品制备,在(超)lgps固态电池中进行1次充电
‑
放电循环之后,将复合阴极和(超)lgps的冷压薄膜从填充氩气的手套箱内取出。然后将其安装到sem短柱上并密封到同一手套箱内的塑料袋中。fib样品制备在fei helios 660双束系统上进行。然后将制备的fib样品立即转移到joel 2010f中,以进行tem和stem eds/eels表征。
[0223]
密度泛函理论计算
[0224]
为了允许与材料项目晶体数据库的可比性,使用材料项目标准进行所有dft计算。所有计算都在vasp中使用推荐的投影缀加平面波(paw)赝势进行。使用k点网格为1000/原
子的520ev的能量截止。通过离散地评价材料在0gpa和1gpa之间的平均压缩率,得到压缩率值。在驰豫和自洽场计算期间,通过对应力张量施加外部应力,计算在各种压力下的焓。
[0225]
实施例3
‑
选择最佳界面涂层的计算方法
[0226]
与液态对应物一样,固态电解质的关键性能度量是稳定性和离子传导率。对于锂系统,两个非常有前景的固态电解质家族是石榴石型氧化物和陶瓷硫化物。这些家族分别由llzo氧化物和lsps硫化物的高性能电解质表示。氧化物倾向于在宽电压范围下维持良好的稳定性,但通常具有较低的离子传导率(<1ms cm
‑1)1。相反地,硫化物可以达到优异的离子传导率(25ms cm
‑1)
6,20
,但是当暴露于电池工作所需的条件时倾向于分解。
[0227]
固态电解质的不稳定性可以由固有材料层面的本体分解或当与其他材料接触时的表面/界面反应引起。在材料层面上,固态电解质倾向于化学稳定(即,自发分解极少),但对与由电池单元形成的锂离子储库的电化学反应敏感。电压稳定窗口限定了锂化学势的范围,在所述范围内固态电解质将不会电化学分解。电压窗口的下限表示还原的开始,或锂离子和相应电子的消耗,而上限表示氧化的开始,或锂离子和电子的产生。当始终经受施加的电压时,电压窗口影响任何固态电解质颗粒的本体。虽然在接触点处固态电解质和第二
‘
涂覆’材料之间发生界面反应,但是这些反应可以是其中仅固态电解质和涂覆材料是反应物的两体化学反应,或者是其中固态电解质、涂覆材料和锂离子储库都参与的三体电化学反应。这两种类型的反应分别是独立于和依赖于充电状态或电压的,这由锂离子储库的参与决定。
[0228]
先前的研究已经揭示,最常见的锂离子电极材料(如licoo2(lco)和lifepo4(lfpo))与大多数固态电解质、特别是高性能陶瓷硫化物形成不稳定的界面。在固态电池中成功地应用陶瓷硫化物可以采用能够减轻这些界面不稳定性的合适的涂覆材料。这些涂覆材料可以是固有电化学稳定的,并且在工作的全电压范围内与陶瓷硫化物形成电化学稳定的界面。另外,如果为了最大材料层面稳定性而在不同的单元组件中使用不同的固态电解质,则涂覆材料也可以改变以维持化学稳定的界面。
[0229]
简而言之,涂覆材料的选择取决于固态电解质的类型和工作电压的预期用途(阳极膜、隔膜、阴极膜等)两者。伪二进制计算方法可以近似地解出给定界面的稳定性,但是计算成本高并且还没有被非常大规模地开发。界面稳定性的高通量分析的主要性能瓶颈是构造和评价许多高维凸包(convex hull)的成本。在材料相稳定性的情况下,问题的维度由元素的数目支配。例如,计算lsps和lco的界面化学稳定性将需要对应于元素集{li,si,p,s,co,o}的6维hull。在系统对锂开放的情况下,计算这种界面的电化学稳定性,从而将锂从所述集中去除,并且所需的hull变成5维的({si,p,s,co,o})。
[0230]
在此,我们引入新的计算方案以更有效地进行界面分析,并且因此能够在考虑固态电解质和工作电压范围两者的情况下有效高通量搜索合适的涂覆材料。我们通过应用这些方案来搜索来自材料项目(mp)的超过67,000个材料条目以找到用于lsps的合适涂覆材料来证明这些方案,所述涂覆材料已经显示在阳极操作和阴极操作两者的情况下,最高锂电导率为约25ms cm
‑1。在材料层面上固有稳定并且在规定电压范围内与lsps形成稳定界面的涂覆材料候选物被称为“功能稳定的”。
[0231]
为了建立标准,我们集中于找到在相对于锂金属0
‑
1.5伏的窗口中功能稳定的阳极涂覆材料和在相对于锂金属2
‑
4伏的窗口中功能稳定的阴极涂覆材料。这些电压范围基
于在当今锂离子电池中通常见到的循环范围。在阳极范围内,我们特别感兴趣的是找到在相对于锂金属0伏下稳定的材料,因为它能够使锂用作市售阳极材料。
[0232]
由于剩余的计算限制,这项工作仅集中于需要lsps界面hull维度小于或等于8的那些材料。换句话说,仅当材料中存在的元素由{li,si,p,s}加上至多四种另外的元素组成时才考虑所述材料。对mp数据库中总共69,640种晶体结构进行材料层面电压窗口的评价。其中,67,062种材料满足小于8维的要求,并且因此用lsps评价功能稳定性。总之,对于lsps,发现超过1,000个mp条目在阳极范围内是功能稳定的,并且超过2,000个mp条目在阴极范围内是功能稳定的。界面稳定性的实验探测用于选择材料以证实这些预测。
[0233]
结果和讨论
[0234]
数据获取和计算效率
[0235]
为了有效地评价这些67,062种潜在涂覆材料中的每一种与lsps之间的界面的稳定性,开发了两种新的计算方案。为了使必须计算的hull数目最少,基于元素组成将涂覆材料分档(bin)。每个独特的元素集需要不同的hull,但是可以同时求解元素子集。例如,lsps和硫酸铁(fe2(so4)3)之间的界面稳定性的计算需要求解6维元素集{li,si,p,s,fe,o}的凸包。这种hull是必须对与lfpo的界面计算的相同hull,并且包括作为子集的评价硫化铁(fes)所需的5维hull。为了利用这一点,不是迭代通过67,062种材料中的每一种并且计算所述材料所需的hull,而是确定跨越全部材料的元素集的最小数目(图25a)。然后,对于每个元素集,仅需要一个hull来评价可以使用那些元素构造的所有材料。这种方法将所需hull总数从67,062个(每种材料一个)减少到11,935个(每个元素集一个)。如图25a中所见,需要维度小于7的少许hull。那些否则将需要低维hull的化合物被求解为较大元素集的子集。另外,由于相同组成空间的多个相需要相同的hull,所以所需的7和8维hull的数目大大减少。
[0236]
用于最小化计算成本的第二方案是用于一旦计算出hull就确定伪二进制的二进制搜索算法。伪二进制方法在图25b中示出。由于在两种材料之间的界面处的分解可以消耗任意量的每种材料,因此所消耗的两种材料之一的分数(在等式1中,x)可以在0
‑
1之间变化。
[0237]
(1
‑
x)lsps+xa
→
∑d
i
d
i
ꢀꢀꢀ
(1)
[0238]
伪二进制是确定由等式1描述的分解的哪个x值是最强动力学驱动的(例如,何时分解能最严重)的计算方法。等式1的rhs表示每种热力学上有利的衰减产物的分数({d
i
}),并且关于产物的gibbs能定义给定x的凸包(hull(x)=∑d
i
(x)g
i
)。伴随等式1的总分解能是:
[0239]
g
hull
(x)=∑d
i
(x)g
i
‑
(1
‑
x)g
lgps
‑
xg
a
ꢀꢀꢀ
(2)
[0240]
lsps和涂覆材料之间的最强动力学驱动的反应是最大化等式2的量值(即,最负)的反应,其定义了参数x
m
。
[0241]
max|g
hull
(x)|≡|g
hull
(x
m
)|
ꢀꢀꢀ
(3)
[0242]
这种最大分解能是两个因素的结果。第一个因素,表示为是由于这两种材料的固有不稳定性而产生的分解能的部分。关于lsps的分解产物(d
lsps
)和涂覆材料的分解产物(d
a
),是对应于反应(1
‑
x)lsps+xa
→
(1
‑
x)d
lsps
+xd
a
的分解能。通过从总hull能量
中减去这种材料层面的不稳定性,可以分离出界面的效应(g
′
hull
),如等式4中所定义。
[0243][0244]
物理上,表示材料在隔开时的不稳定性,并且g
′
hull
(x)表示一旦使材料接触,则由界面引起的不稳定性的增加。
[0245]
在这项工作中,为了确定在最强动力学驱动的分数下每个界面的增加的不稳定性(g
′
(x
m
)),我们实施了使用hull的凹度以在0.01%误差内找到x
m
的二进制搜索算法(参见“方法”)。这种二进制搜索方法在14步的hull评价中找到x
m
值。以0.01%的精度对hull的更传统线性评价将需要从x=0到x=1的10,000次等距评价。这种速度的增加被用来有效地搜索67,062个材料条目的功能稳定性。
[0246]
功能稳定性
[0247]
通过如下要求,确定67,062种材料中的每一种在给定电压下的功能稳定性:(i)在所述电压下材料的每原子固有电化学稳定性低于热能(|g
hull
(x=1)|≤k
b
t),和(ii)在给定电压下增加的界面不稳定性低于热能(|g
′
hull
(x
m
)|≤k
b
t)。在这些条件下,系统中的唯一不稳定性是lsps固有材料层面不稳定性,其可以通过应变诱导方法
22
来稳定。在67k种材料中,发现1,053种材料在阳极范围(相对于锂金属0
‑
1.5v)是功能稳定的,并且发现2,669种材料在阴极范围(相对于锂金属2
‑
4v)是功能稳定的。另外,确定152种材料在阳极范围中和142种材料在阴极范围中违反条件(i),但仅通过锂化/脱锂来分解。这种材料作为lsps涂覆材料的实际使用取决于这种锂化/脱锂过程的可逆性,因此这些材料被称为潜在功能稳定的。所有功能稳定和潜在功能稳定的材料都编目在补充信息中,并由相应的材料项目(mp)id索引。
[0248]
每种元素的原子分数和界面稳定性之间的相关性描绘在图25c和图26a
‑
26c。图25c描绘化学反应中每种元素与g
′
hull
(x
m
)的相关性,而图26a
‑
26c分别描绘在相对于锂金属0v、2v和4v下的电化学反应中每种元素与g
′
hull
(x
m
)的相关性。元素组成和g
′
hull
(x
m
)之间的负相关性意味着增加该元素的含量改进界面稳定性。图25c指示对于那些含有大阴离子(如硫、硒和碘)的化合物,化学稳定性是最好的。总体上,图26a和图26c分别指示在低电压和高电压下在元素物种和g
′
hull
(x
m
)之间存在降低的相关性。这提示在这些电压极值处,界面分解由固有材料层面的还原/氧化而不是界面效应(g
′
hull
)主导。在相对于锂的2v(图26b)下,对于大多数元素看到正相关性(较高的不稳定性),其中显著例外的是硫属元素和卤素阴离子基团,它们是负相关的。
[0249]
阴离子物种对材料层面稳定性的影响
[0250]
考虑到阴离子物种相对于界面稳定性的高相关性对比,进行关于阴离子组成的数据集的分析。为了消除数据点之间的重叠,所考虑的仅有化合物是仅具有{n,p,o,s,se,f,i}中的一个的单阴离子或具有氧加{n,s,p}中的一个的氧
‑
阴离子的那些。45,580个mp条目满足这些标准之一,如表3中所概述。还提供了发现在材料层面电化学稳定的每种阴离子类别的百分比。
[0251]
表3.单阴离子和氧
‑
阴离子数据集的大小以及在阳极范围(0
‑
1.5v)和阴极范围(2
‑
4v)中电化学稳定的每种的百分比。例如,f表示在化学式中含有f的所有化合物,而o+n表示在化学式中含有o和n两者的所有化合物。
[0252][0253]
图27a示出施加的电压对材料(在这种情况下,lsps)的hull能量的影响。当hull能量相对于电压的斜率为负时,相应的分解是还原,而如果所述斜率为正,则相应的分解是氧化。在中间,存在其中hull斜率为零的区域,这意味着不存在与锂离子储库的反应(即,所述反应相对于锂是中性的)。考虑到这一点,图27b和图27c分别绘制每种阴离子类别在阳极范围和阴极范围内的特征性氧化还原行为。在45
°
的“中性衰减”线表示在两个电压极值下具有相同hull能量并且因此不与锂离子反应的那些化合物。在该线之上[之下]的数据点是hull能量相对于电压增加的[降低的],并且因此在所绘制的电压范围内是特征性氧化性[还原性]的。
[0254]
图27b指示,与预期一致,大多数化合物在相对于锂金属0
‑
1.5v的阳极电压范围内被还原。可以看出含氮化合物不成比例地占据y轴,指示当与锂金属直接接触时具有更高的稳定性水平。这与先前的计算工作一致,先前的计算工作指示二元氮化物和三元氮化物对锂金属比硫化物或氧化物更稳定
33
。然而,在阴极电压范围内(图27c),看到阴离子类别的变化大得多。含氧
‑
阴离子和氟的化合物主要保持还原性,而含磷、硫化物和硒的化合物的特征是氧化性的。含氧化合物在中性衰减线的两侧上都见到,这意味着氧化物可能在这种2
‑
4v范围内锂化/脱锂。
[0255]
在图27d中,每种阴离子类别的平均hull能量自0
‑
5v以0.5v步长给出。含氮化合物被证实在0v时最稳定,而碘和磷化合物维持相当的稳定性。对于0.5v和1.0v之上的电压,磷和碘的平均稳定性分别超过氮。在高电压(>4v)下,可以看出含氟和碘的化合物是稳定的,而含氮化合物是最不稳定的。
[0256]
阴离子物种对界面层面稳定性的影响
[0257]
对于每种阴离子类别,总分解能的平均值(g
hull
(x
m
))和作为界面不稳定性结果的分数(g
′
hull
(x
m
))描绘在图28a
‑
28c中。图28a显示由于阴离子类别和lsps之间的化学反应而引起的平均不稳定性。含硫和硒的化合物平均形成与lsps的最化学惰性的界面。相反地,含氟和氧的化合物是最具反应性的。作为一般趋势,在总项(较高的g
hull
(x
m
))中更不稳定的那些化合物类别也维持相对于固有材料贡献更高的界面贡献(g
′
hull
(x
m
))。这意味着在决定界面的化学稳定性方面,每种类别的固有化学稳定性的差异所起的作用不如其与lsps的反应性显著。
[0258]
图28b显示界面自0
‑
5v以0.5v步长的平均总电化学分解能。总体上,每种阴离子类别遵循似乎由lsps的材料层面电化学稳定性主导的路径(图27a)。在低电压状态(<1v)和高电压状态(>4v)下尤其如此,在这些状态下电化学效应将是最为显著的。界面稳定性与lsps的固有稳定性的最大偏离发生在1
‑
3v的区域。在这种“中”电压范围内,具有最低化学
分解能的那些化合物(含有s、se、i、p的化合物)与lsps的偏离最小,而具有大分解能的那些化合物(含有n、f、o、o
+
的化合物)偏离更显著。这种趋势提示,低电压范围和高电压范围分别由材料层面的电化学还原和氧化主导,而中范围由界面层面的化学反应主导。例如,在0v下,预期在al2o3和lsps之间的界面衰减成{li9al4,li2o,li3p,li2s,li
21
si5},其是将由在0v下独立分解的每种材料产生的同一组衰减产物。因此,界面的存在没有能量效应。
[0259]
电化学分解的平均界面层面贡献示于图28c中。所有阴离子类别在0v下都趋向于g
′
hull
(x
m
)=0,这意味着材料在0v下趋向于变得完全被还原,在这种情况下,与材料层面的不稳定性相比,界面效应是可忽略的。在中电压范围内出现明显的界面不稳定性,而在高电压范围内界面不稳定性再次降低。同样,这意味着界面层面化学效应在中电压范围内占优势,而材料层面还原[氧化]在低[高]电压下占优势。在高电压下,界面对不稳定性的贡献接近最大程度氧化的材料与lsps之间的反应能。结果,对于任何高于4v的电压,界面将增加能量不稳定性到与这种化学反应相等。这解释了高电压渐近行为,而低电压行为总是趋向于0ev原子
‑1。例如,对于任何高于4v的电压,lfpo将分解为{li,fepo4}},而lsps将分解为{li,p2s5,sis2,s}。界面的引入允许这些氧化产物发生化学反应并形成fes2和sio2。
[0260]
阴离子物种对功能稳定性的影响
[0261]
图29a(阳极范围)和图29b(阴极范围)中给出被确定为功能稳定或潜在功能稳定的每种阴离子类别的总数,其中它们在材料层面上是固有稳定的,并且在规定的电压范围内与lsps形成稳定的界面。对于阳极范围,含氮、磷和碘的化合物具有最高百分比的稳定化合物(2
‑
4%),而所有其他类别低于1%。阴极范围显示高得多的百分比,其中含硫化合物达到35%。碘和硒两者都高于10%。
[0262]
实验比较
[0263]
通过手工研磨lsps和涂覆材料的混合物粉末,进行/不进行高温退火,然后在室温下进行x射线衍射(xrd)测量,实验性地测试各种涂覆材料和lsps之间的化学相容性。粉末之间的任何化学反应都将引起原始相中的组成和结构变化,这可以通过xrd图谱中的峰位置和强度的变化来检测。值得注意的是,即使基于热力学计算预测界面反应会发生,也可能需要一定量的能量来克服这些反应发生的动力能垒4。因此,将混合粉末在高温(300℃、400℃、500℃)下退火以确定界面反应的起始温度以及反应产物,并且通过将这些结果与dft计算的热力学反应产物进行比较来进一步评估动力学的作用。
[0264]
图30a
‑
30d比较了这种室温和500℃退火的粉末混合物的xrd图谱。将几种候选涂覆材料(即sno2、li4ti5o
12
、sio2)与lsps混合(图30c
‑
30d),而lco+lsps的混合粉末用于比较(图30a)。将在室温和500℃下的每个单个相(即sno2、li4ti5o
12
、licoo2、sio2和lsps)的xrd图谱用作参考(图31a
‑
31e)。通过比较这些xrd图谱,很明显,在室温下,没有涂覆材料与lsps反应,因为xrd图谱仅显示原始相的峰。然而,在500℃下退火6小时后,不同的材料显示与lsps完全不同的反应能力。观察到lco与lsps严重反应,因为混合粉末的xrd图谱的峰强度和位置在10
‑
80度的整个2
‑
θ范围内完全改变(图309a)。原始lco和lsps峰消失或减少,而出现属于新反应产物(如sio2、li3po4、立方co4s3和单斜co4s3)的额外峰,指示lco与lsps不相容。作为鲜明对比,sio2+lsps混合物的xrd图谱的峰强度和位置从未改变,在500℃退火之前和之后都仅显示原始峰。这是显示当sio2与lsps接触时没有界面反应发生的直接证据,尽管提供了大量外部能量。sno2和lto也显示出与lsps不相容,因为在其500℃退火的样
品的xrd图谱中出现属于反应产物的新峰,然而,反应产物的峰比lco+lsps的情况弱得多。在图30a
‑
30d中通过颜色区域突出显示了四种材料的峰位置和强度变化的2
‑
θ范围,作为不同材料与lsps不相容的指示。从图30a
‑
30d可以观察到,这种不相容性顺序是lco>sno2>lto>sio2,这与我们基于热力学计算的理论预测完全一致。各种材料与lsps的界面反应的起始温度示于图32a
‑
32d中。
[0265]
典型的涂覆材料的电化学稳定性通过循环伏安法(cv)技术来表征,其中所测试的涂覆材料的分解可以通过在相对于锂的某些电压下的电流峰来显现。将两种典型的涂覆材料用作展示,以显示我们的理论预测和实验观察之间的良好对应性。li2s的cv测试(图30e)显示在0
‑
1.5v之间的相对平坦的区域,而大的氧化峰在2
‑
4v的区域占优势。相反,sio2的cv测试(图30f)展示在0
‑
1.5v的区域的净减少,并且在2v和4v之间存在具有很少分解的中性区域。这些结果同样是证实我们基于热力学计算的理论预测的直接证据。
[0266]
方法
[0267]
数据采集
[0268]
这项工作中使用的数据是先前密度泛函理论计算的结果,所述先前密度泛函理论计算作为材料项目(mp)的一部分来执行,并且使用材料应用编程接口(api)来与之连接。使用python材料基因组学(pymatgen)库来计算凸包。在被评价的初始69,640种结构中,2,578种结构由于要求hull的维度等于或大于9,因此没有考虑。
[0269]
元素集送代
[0270]
为了最小化分析所有67,062种结构的计算成本,确定跨越所有材料的元素集的最小数目。为此,将每种结构中的元素集与lsps的元素组合,产生元素集的列表,其中各集合的长度等于该材料的所需hull的维度。这个列表基于集合的递减长度来排序(例如,以所需hull的递减维度来排序)。然后,迭代这种集合,并且去除等于前一集合或者是前一集合的子集的任何集合。结果是元素集的最小数目,其中可以描述每种材料。
[0271]
使用来自mp的能量和组成计算化学分解hull。忽略体积和熵的变化(δg≈δe)。相似地,通过使用锂巨正则自由能并从能量中减去项μ
li
n
li
(δφ≈δe
‑
μ
li
δn
li
)得到电化学分解hull,其中μ
li
是目标化学势并且n
li
是结构中锂离子的数目。在计算出hull之后,使用其来评价存在于其元素集的跨度内的每一种材料。
[0272]
伪二进制
[0273]
如第2节中所述,伪二进制寻求找到lsps与涂覆材料的比率,以使得分解能最严重,并且因此是最强动力学驱动的。通过使用向量符号以通过将原子占位映射到向量元素来表示给定的组成,简化了这个问题。例如,以(li co o)为基础,licoo2→
(112)是指在单元式中存在1个锂、1个钴和2个氧。使用这种符号,可以以向量形式写出等式1中的分解。
[0274][0275]
使用来表示向量和表示矩阵,等式5变为:
[0276][0277]
每种衰减产物的相对组成导数可以通过在等式6中对求逆来得到。
[0278][0279]
等式7允许计算hull能量相对于分数参数x的导数。
[0280][0281]
通过使用等式7、以及该hull是其x的凸函数的事实,可以执行二进制搜索以找到g
hull
的最大值及其出现x
m
时的值。这个过程由以下组成:首先定义一个二元素向量,其限定其中已知存在x
m
的范围x
范围
=(0,1)和初始猜测x0=0.5的范围。在初始猜测下评价凸包产生分解产物{d
i
}和相应的能量然后,可以使用等式7和等式8来得到hull能量的斜率。如果hull能量是正的,则x
范围
→
(x0,1),而如果它是负的,则x
范围
→
(0,x0)。重复这个过程,直到上限和下限相差小于0.01%的规定阈值的因子,这将总是在14个步骤中实现(2
‑
14
≈0.006%)。
[0282]
等式5
‑
8是针对化学稳定性定义的。在电化学(锂开路)稳定性的情况下,自由能被φ
i
=g
i
‑
μn
i
代替,其中μ为化学势且n
i
为结构i中锂的数目。另外,锂组成不包括在等式6的组成向量中,以允许锂原子的数目改变。
[0283]
x射线衍射
[0284]
通过xrd在室温(rt)下研究候选材料和固态电解质的相容性。xrd样品通过在填充ar的手套箱中手工研磨候选材料(lco、sno2、sio2、lto)与lsps粉末(重量比=55∶30)来制备。为了测试候选材料和lsps固态电解质的反应的起始温度,将粉末混合物充分铺展在加热板上以加热至不同的标称温度(300℃、400℃和500℃),然后通过xrd表征。
[0285]
xrd测试在配备有在10
‑
80
°
的2
‑
θ范围内的cu kα辐射的rigaku miniflex 600衍射仪上进行。所有xrd样品架都在填充ar的手套箱中用kapton膜密封以避免在测试期间暴露于空气。
[0286]
循环伏安法
[0287]
将候选涂覆材料(li2s和sio2)、炭黑和聚(四氟乙烯)(ptfe)以90∶5∶5的重量比混合在一起,并在填充ar的手套箱中手工研磨。将粉末混合物顺序地手工滚压成薄膜,从其中冲压出圆盘(直径5/16英寸,~1
‑
2mg装载量)以形成用于循环伏安法(cv)测试的工作电极。将这些电极组装成swagelok单元,其具有作为对电极的li金属、两个玻璃纤维隔膜和市售电解质(在1∶1(体积比)碳酸亚乙酯/碳酸二甲酯(ec/dmc)溶剂中的1m lipf6)。
[0288]
cv测试由solartron 1455a以0.1mv/s的电压扫掠速率在0
‑
5v的范围内在室温下进行,以研究候选涂覆材料(li2s和sio2)的电化学稳定窗口。
[0289]
结论
[0290]
我们对材料项目dft数据的高通量伪二进制分析已经揭示,与lsps的界面在1.5v至3.5v的范围内主要通过化学方式以及在更低[更高]电压下通过电化学还原[氧化]而衰减。当电压接近0v时,归因于界面效应的分解能的分数消失。这种结果提示,所有材料类别倾向于在低电压下衰减至最大程度锂化的li二元和元素化合物,在这种情况下,界面的存在没有影响。
[0291]
关于阴离子含量,我们看到,使工作条件与涂覆材料适当匹配是最重要的。例如,含硫和硒的化合物在2
‑
4v阴极范围内展示非常高的功能稳定的机会(在所有硫化物和硒化
物中,>25%)。然而,小于1%的这些相同材料在0
‑
1.5v阳极范围内形成功能稳定的涂覆材料,其中碘、磷和氮具有最高性能。含氧化合物具有在两个电压区域中都功能稳定的大量相,但是由于含氧数据点的数目甚至更高,所以百分比较低。
[0292]
实施例4
[0293]
我们表明,先进的机械压缩方法可以改进在lgps作为电解质的固态电池中锂金属阳极的稳定性。更重要地,我们证明在对称电池中,即使在10ma cm
‑2下进行高倍率测试之后,也不存在li枝晶形成和穿透。机械压缩方法在技术上通过在电池单元上施加100mpa至250mpa的外部压力来实现,其中li金属阳极被石墨膜(g)覆盖,所述石墨膜(g)隔开电池组装件中的lgps电解质层。在最佳li/g容量比下,其在li/g
‑
lgps
‑
g/li对称电池和li/g
‑
lgps
‑
licoo2(linbo3涂覆的)电池两者中都表现出优异的循环性能。在循环时,li/g阳极从两层转变成一个集成的复合层。密度泛函理论(dft)数据与x射线光电子能谱(xps)分析之间的比较产生控制lgps分解反应的机械压缩的首次直接观察。此外,在最佳的压缩条件下,可以看到分解程度受到显著抑制。
[0294]
li/石墨阳极的设计
[0295]
我们首先通过在填充氩气的手套箱内在500℃下对lgps和(锂化)石墨的混合物进行36小时的高温处理以进行加速反应,来研究lgps和(锂化)石墨之间的化学稳定性。在热处理之前和之后对不同的混合物进行xrd测量,如图33(a、b、c)中所示。观察到与锂接触的lgps的严重分解,伴随着li2s、ges2和li5gep3的形成(图33a)。相反,如图33b中所示,在加热后,lgps和石墨的混合物没有发生峰变化,证明石墨与lgps在化学上是稳定的。在加热li和石墨粉末的混合物之后,合成锂化石墨(图38)。当锂化石墨进一步与lgps混合时,它是化学稳定的,如图33c中所示,26
°
峰仅有轻微的强度变化。
[0296]
设计li/石墨阳极,如图33(d)中所示。保护性石墨膜通过将石墨粉末与聚ptfe混合,然后覆盖到锂金属上而制成。将li/石墨、电解质和阴极膜三层依次堆叠在一起,然后机械压制。在电池测试期间,压力维持在100
‑
250mpa下。基于本领域的常识,这种压力有助于获得阳极和电解质之间的良好接触,但更重要地,其起到机械压缩的作用,以改进固态电解质的电化学稳定性。扫描电子显微镜(sem)显示石墨颗粒在如此高的压力下转变成致密层(图39)。在图33e、图33f中,在电池测试之前如此制备的阳极可以通过sem和聚焦离子束(fib)
‑
sem直接观察。清楚看到li、石墨和lgps三层的紧密界面接触。
[0297]
li/石墨阳极的循环和倍率性能
[0298]
用阳极
‑
lgps
‑
阳极对称电池设计,在100mpa的外部压力下,测试li/石墨(li/g)阳极的电化学稳定性和倍率性能。li/g
‑
lgps
‑
g/li电池和li
‑
lgps
‑
li电池之间的循环性能的比较示于图34a中。li对称电池在失效前在0.25ma cm
‑2的电流密度下仅工作10小时,而li/g对称电池在循环500小时之后仍运行,其中过电压缓慢增加至0.28v。如图40中所示,根据在300小时的循环之后过电压从0.13v缓慢增加至0.19v的另一电池,稳定的循环性能是可重复的,指示这种轻微的过电压变化由电池组装件而不同。sem显示,li/石墨阳极在长期循环之后从两层转变为一个集成的复合层,而总厚度没有显著变化(图41)。在图34b中,将在对称电池中在300小时循环之后li/g阳极的sem图像与10小时循环之后li阳极的sem图像相比较。在长期循环之后li/g阳极维持锂/石墨复合材料的致密层(图34b1、图34b2)。相比之下,在10小时的测试之后,在li阳极中出现无数的孔隙,这很大可能是由lgps与li金属的严重
分解反应诱导的。所述孔隙对离子传导率和电子传导率两者都有害,其可能是造成10小时时急剧电压增加的原因,此时li对称电池失效。
[0299]
我们还比较了li/g对称电池在100mpa或3mpa的不同外部压力下的倍率性能,如图34c中所示。通过改变每次循环的工作时间,为不同的电流密度设定相同的充电和放电容量。li/g对称电池可以稳定地从0.25ma cm
‑2循环至3ma cm
‑2,其中过电压从0.1v增加至0.4v。然后其可正常地循环回到0.25ma cm
‑2(图34c1)。而在3mpa下,电池在测试期间在2ma cm
‑2下失效(图34c2)。注意,在相同的电流密度下,100mpa下的过电压仅为3mpa下的过电压的约63%。在直至2ma cm
‑2的倍率测试之后,li/g
‑
lgps界面的sem图像显示在100mpa下紧密的界面接触(图34d1),而在3mpa下测试之后观察到裂纹和空隙(图34d2)。因此,外部压力在电池测试期间起到维持紧密界面接触的作用,从而有助于更好的倍率性能。
[0300]
为了进一步理解通过电池循环形成的li/g复合材料对其高倍率性能的影响,设计了类似于图34(e1)的电池测试。在此,在测试期间保持250mpa的较高外部压力。它在0.25ma cm
‑2下开始1次循环,然后直接达到5ma cm
‑2充电,其显示导致安全停止的急剧增加的电压。然后,我们立即重新启动电池,再次以0.25ma cm
‑2运行十次循环,接着以5ma cm
‑2运行接下来的十次循环。这一次,电池正常地在5ma cm
‑2下运行,平均过电压为0.6v,并且它仍然可以返回到在0.25ma cm
‑2下的循环,而没有明显的过电压增加。在固定电流下,在5ma cm
‑2下的初始电压浪涌指示电阻跳变,这最可能与li和石墨在组装时是两层的事实有关,并且因此在石墨中没有足够的li来支持这样的高电流密度。然而,在0.25ma cm
‑2下循环20小时之后,li/g开始转变成复合材料,如图34b和图41中所示,其中更多的li存储以支持高倍率循环测试。
[0301]
基于上述理解,我们进一步将初始循环的电流密度降低至0.125ma cm
‑2,并且以0.25mah cm
‑2的相同容量循环,以获得更均匀的li分布和在li/g复合材料中的存储,从而改进锂转移动力学。如图34(e2)中所示,电池可以在10ma cm
‑2的电流密度下循环,并且当电流密度设定回到0.25ma cm
‑2时正常循环。注意,在高倍率测试之前和之后,在相同的低电流倍率下没有明显的过电压增加,如图34e和图42的插图所示,其中这一电池的li/g阳极的sem也显示出li/g复合材料的清楚形成,在界面上没有观察到明显的li枝晶。
[0302]
全固态电池中的li/石墨阳极
[0303]
我们首先在相对于锂金属0.0
‑
2.2v的低电压范围内进行lgps分解途径的dft模拟。材料层面上的机械压缩由系统的有效本体模量(k
eff
)来参数化。基于这一模量的值,系统可以从等压(k
eff
=0)到等容(k
eff
=∞)变动。实际电池系统中kefr的预期值为15gpa的数量级。下面,将这些模拟结果用于解释在cv、倍率和循环测试之后,在固态电池中来自lgps的ge和p的化合价变化的xps结果。
[0304]
如图36a中所示,在高效模量下lgps的分解能力较低,指示lgps在低电压下的分解很大程度上被机械压缩所抑制。预测的分解产物和分数值分别列于图36b和表4中。在k
eff
=0gpa(即,没有施加机械约束/等压)时,当电压接近零时,还原产物接近锂二元化合物li2s、li3p和li
15
ge4。然而,在施加机械压缩并且将有效模量设定在15gpa下之后,ge元素、li
x
p
y
和li
x
ge
y
的形成受到遏制,而化合物如p
x
ge
y
、ges和p2s出现。这也与已知p
x
ge
y
是高压相的事实一致。图36中所示的在不同k
eff
下的电压分布和还原产物指示在施加机械压缩之后,lgps的分解在低电压下遵循不同的还原途径。
[0305]
表4.(a)
‑
(d)在不同k
eff
下降至低电压的lgps分解产物与分数值
[0306]
(a)k
eff
=0gpa
[0307][0308][0309]
(b)k
eff
=5gpa
[0310][0311]
(c)k
eff
=10gpa
[0312][0313][0314]
(d)k
eff
=15gpa
[0315][0316]
值得注意的是,虽然施加的压力和有效模量(k
eff
)两者均以压力为单位测量,它们是独立的。有效模量表示与电池系统的有限刚性平行地增加的电解质的固有体积模量。因此,k
eff
测量可以在任何单个颗粒中在材料层面上实现的机械压缩,而对固态电池的工作施加的外部压力加强这种压缩对颗粒之间或电极和电解质层之间的界面上的有效性。这是因为暴露的表面最易受化学和电化学分解的影响,而由外部压力所施加的紧密界面接触将使这种表面最小化。因此,即使施加的压力仅在100mpa的数量级,也预期有效本体模量会大得多。事实上,密堆积的lgps颗粒将经历大约15gpa的k
eff
。100
‑
250mpa的施加压力对获得这种密堆积结构是有效的工具。简而言之,施加的压力使本体电解质中的间隙最小化,允许表示对材料层面的机械压缩的有效模量接近其约15gpa的理想值。
[0317]
在电池循环期间与锂或锂
‑
石墨阳极直接接触的lgps以及本体lgps的xps结果提供在图37中。根据图36b的相预测,可以很好地理解这些化合价变化的测量。远离阳极界面的隔膜区域中的lgps显示出与原始lgps相同的ge峰和p峰(图37a)。
[0318]
我们首先研究与纯锂金属相比较,li/g复合材料在0.25ma/cm2的慢倍率下在100mpa外部压力下的功能(图37b,图37c)。使用纯锂金属(图37c),在li
‑
lgps界面上ge和p两者的还原都是显著的,表明形成li
x
ge
y
合金、元素ge和li3p。注意,li
x
ge
y
中的ge化合价和li3p中的p化合价为负或低于零价,与来自dft模拟的bader电荷分析一致(图44)。相反,使用li/g阳极,还原在li/g
‑
lgps界面上受到抑制,在分解的化合物中ge和p的化合价都保持高于零(图37b)。li和lgps界面是化学不稳定的,导致包括图37c中观察到的化合物的分解。这些分解也与图36b中在0gpa的k
eff
下预测的分解一致。这种化学分解的界面的进一步电化学循环将导致分解的体积分数增加,最终消耗所有的lgps。相反,li/g阳极中的石墨层阻止在lgps和li之间的化学界面反应,而在适当的机械压缩下,电化学分解似乎通过在图36b中
高k
eff
≥10gpa的路径,其中ges、p
x
ge、p2s与图37b中自xps观察的化合价匹配。
[0319]
当循环倍率增加到2ma/cm2和10ma/cm2时,在图37d、图37e中在外部压力下在l/g
‑
lgps界面上观察到的分解改变为亚稳途径,其不同于图37b中在0.25ma/cm2下的低倍率途径。这意味着虽然图37b与图36中预测的热力学一致,但在高电流密度下分解变为动力学主导的。此外,可以得出结论,在图37d、图37e中所见的li/ge合金形成,代替还原的p,是动力学优先的相。具体地,基于来自xps的化合价,ge0和li
x
ge
y
以及li3ps4和li7ps6最可能分解。注意,在3mpa的外部压力下且因此在界面上降低的k
eff
下,即使在2ma/cm2的高倍率下,也观察到ge和p两者的还原(图37f),与图36b中在低k
eff
下预测的一般趋势一致。然而,p还原可能仍然是动力学速率限制的,因为没有观察到li3p的最被还原态,如在图36b中在k
eff
=0gpa下预测的和如在图37c中从界面化学反应观察到的。
[0320]
这两个分别为热力学优先和动力学优先的竞争反应可以通过考虑这两个竞争反应(η
→
η+η
′
(i))中每一个的电流依赖性过电压(η
′
(i))来理解。该η
′
项将由动力学效应(如欧姆损耗等)引起。当电流为小(i≈0)时,η
′
消失,因此热力学过电压(η)主导并有利于图36的基态分解产物。然而,在高电流下,η
′
开始主导并且有利于那些亚稳相,例如在高k
eff
下的li
x
ge
y
,在我们的计算中,其未在图36中示出,因为它们在每个电压范围中都是基态相。
[0321]
在100mpa或3mpa下的cv测试之前和之后的阻抗分布(图45a)在用图45d中所示的模型拟合之后在图45b和图45c中进行比较。计算的r
本体
(本体电阻)和r
ct
(电荷转移电阻,在此主要为界面电阻)列于表5中。由于在高压下的更好接触,在100mpa下的r
ct
(38.8ω)比在3mpa下的r
ct
(395.4ω)小得多。在cv测试之后,100mpa下电池的r
本体
几乎没有任何变化,而在3mpa下电池的r
本体
从300ω增加到600q。显著提高的电阻归因于lgps在无效机械压缩下的更严重的分解。再次,根据电化学测试,证明在最佳压缩条件下分解程度受到显著抑制。
[0322]
表5.计算的r
本体
和r
ct
[0323][0324][0325]
结论
[0326]
锂
‑
石墨复合材料允许在以lgps作为电解质的固态电池的测试期间施加高外部压力。这在材料层面上产生高机械压缩,这有助于li/g
‑
lgps
‑
g/li对称电池的优异倍率性能。对于这种固态电池,在直至10ma cm
‑2的高电流密度下循环之后,循环仍可以正常地以低倍率进行,提示没有锂枝晶穿透或短路。通过使用实验xps测量和dft计算模拟两者来分析在不同机械压缩下lgps分解的还原途径。首次表明,在适当的机械约束下,lgps还原遵循不同的途径。然而,这种途径可能在动力学上受高电流密度诱导的过电压影响。因此,lgps的分解是机械压缩和电流密度两者的函数。根据电池循环性能和阻抗测试,表明高机械压缩以及动力学限制的分解途径降低了总阻抗,并且实现了具有优异倍率性能的lgps
‑
锂金属电
池。
[0327]
方法
[0328]
电化学
[0329]
石墨薄膜是通过将活性材料与ptfe混合而制成的。石墨膜的重量比为石墨:ptfe=95∶5。所有的电池都是使用自制的加压单元在氧气和水<0.1ppm的填充氩气的手套箱中组装的。对称电池(li/g
‑
lgps
‑
g/li或li
‑
lgps
‑
li)通过将li(/石墨)
‑
lgps粉末
‑
(石墨/)li三层冷压在一起而制成,并在电池测试期间保持在不同的压力下。电池在不同的电流密度下充电和放电,每次循环的总容量为0.25mah cm
‑2。licoo2半电池通过用液压机冷压li/石墨复合材料
‑
lgps粉末
‑
阴极膜而制成,且保持压力在100
‑
250mpa下。licoo2使用溶胶
‑
凝胶法用linbo3涂覆。所有阴极膜的重量比为活性材料:lgps:ptfe=68∶29∶3。在land电池测试系统上获得电池循环数据。在25℃下在0.1c下测试循环性能。cv测试(li/g
‑
lgps
‑
lgps/c)在solartron 1400单元测试系统上在ocv至0.1v之间以0.1mv/s的扫描速率进行。用于cv测试的lgps阴极膜由lgps:super p:ptfe=87∶10∶3制成。
[0330]
材料表征
[0331]
xrd:xrd样品通过在手套箱中以重量比=1∶1将lgps粉末与锂金属和/或石墨手工研磨来制备。将粉末混合物放在加热板上并加热至标称温度(500℃)持续36小时,然后通过xrd表征。xrd数据使用rigaku miniflex 6g获得。在填充氩气的手套箱中用kapton膜密封高温处理之前和之后的lgps和石墨的混合物,以防止空气污染。
[0332]
sem和xps:li/石墨
‑
lgps
‑
石墨
‑
li的球粒的横截面成像通过supra 55sem获得。将球粒破碎成小块,并用碳带附接在螺母的侧面上,使其垂直于光束。将带有样品的螺母安装到标准sem短柱上,并密封到填充氩气的手套箱内的两个塑料袋中。在feihelios 660双光束系统上进行fib
‑
sem成像。自thermo scientific k
‑
α+获得xps。将样品安装到标准xps样品架上,并且也用塑料袋密封。所有样品在约10秒内转移到真空环境中。所有xps结果都经由avantage通过峰值微分和模拟来拟合。
[0333]
计算方法所有dft计算都使用vienna从头模拟包(vasp)按照材料项目计算参数进行。
32
使用1000kppa的k点密度、520ev的截止值和vasp推荐的赝势。使用如参考文献13中概述的lagrange最小化方案,对于0gpa、5gpa、10gpa和15gpa的有效模量计算机械约束的相位图。考虑材料项目数据库中的所有li
‑
ge
‑
p
‑
s相。bader电荷分析和自旋极化计算用于确定电荷化合价。
[0334]
实施例5
‑
[0335]
在本项工作中,我们集中于如何通过在单元层面上控制,将外部施加的高压或等容条件用于在材料层面上稳定lgps。这种进步超过在先前工作中存在的微观结构层面的机械约束,在所述先前工作中使用颗粒涂层来诱导亚稳性。在适当的机械条件下,我们表明lgps的稳定窗口可以拓宽到9.8v的工具测试上限。测量lgps在高电压保持之前和之后的结构变化的同步辐射x射线衍射(xrd)和x射线吸收光谱(xas)首次显示lgps在这些电化学过程期间应变的直接证据。通过比较在电压稳定窗口之外进行分解分析的密度泛函理论(dft)模拟和x射线光电子能谱(xps)测量,进一步考虑了热力学因素和动力学因素两者。这些结果提示机械诱导的亚稳性将lgps稳定直至大约4v。另外,从4
‑
10v,通过在刚性机械约束中的分解而经历的局部应力导致动力学稳定性。组合的机械诱导的亚稳性和动力学稳定
性允许电压窗口从2.1v扩展到接近10v。为了证明这种方法对于实际电池系统的效用,我们使用这种方法用各种阴极材料构建全固态电池。li4ti5o
12
(lto)阳极与lico
0.5
mn
1.5
o4(lcmo)、lini
0.5
mn
1.5
o4(lnmo)和licoo2(lco)阴极配对,以证明受约束的lgps的高电压稳定性。为了进一步探测lgps的电化学窗口,我们报道了基于锂金属和lico
0.5
mn
1.5
o4的首个全固态电池,其可以充电至6
‑
9v并循环至5.5v。
[0336]
结果
[0337]
为了说明机械约束如何影响lgps的电化学稳定性,进行lgps+c/lgps/li单元的循环伏安法(cv)测试(图46a)。在组装件中用1吨、3吨或6吨(t)的力(分别为78mpa、233mpa和467mpa)预压制三个电池,然后在正常swagelok电池中测试。将紧固的swagelok电池的外部压力校准为数mpa,给出准等压电池测试条件。另外,一个电池初始在6t下压制,然后紧固在自制的加压单元中,在电池测试期间恒定施加的外部压力被校准为约200mpa,施加准等容电池测试环境。在1t、3t和6t下预压制之后lgps球粒的密度分别为单晶lgps理论密度的62%、69%和81%。压制之后lgps球粒的形态示于图51a中。然而,由原位力
‑
位移测量计算的在加压单元中的球粒的密度(图51b)在超过30mpa外部压力下已经接近100%。
[0338]
如图46a中所示,在循环伏安法(cv)测试中,存在阈值电压,每个单元在超过所述阈值电压下开始严重分解。对于在1t、3t和6t下预压制的那些等压单元,这些阈值分别为4.5v、5v和5.8v。然而,将等容单元充电至9.8v,并且没有显示明显的分解。在低电压区域(图46b),对于等压单元,在~3v和~3.6v下可以看到两个较小的分解峰,其中在预压制步骤中在增加的压力下观察到降低的峰强度。相反,等容单元完全避免了这些峰。在cv测试期间,在不同电压下通过阻抗谱测量这四个单元中的电池的原位电阻(图46c)。在此发现预压制中的较高压力改进颗粒之间的接触,并且因此降低了固态电池系统中的初始电阻(在3v下,在图46c中)。然而,当cv测试向高电压进行时,等压单元中的电阻增加得更快,指示阴极中的lgps在弱机械压缩条件下经历了一定的分解。相反,使用等容单元测试的电池的电阻几乎没有变化。值得注意的是,结晶lgps朝向高电压的电压稳定窗口通过机械压缩诱导的亚稳性而从2.1v扩展到约4.0v,在图46a中在电池中观察到远超过4v的5v至10v的稳定性提示了不同的现象。
[0339]
如图46d所示,来自等容单元的lgps的同步辐射xrd指示在cv测试直至9.8v之后lgps的一般晶体结构保持不变。然而,在7.5v和10v下进行高电压cv扫描之后观察到xrd峰加宽(图46e和图52)。发现随着2θ角度增加的峰加宽(图46f)遵循应变加宽机制而不是尺寸加宽。注意,在3.2v处没有观察到明显的应变加宽。
[0340]
这种应变效应从xas测量和分析中进一步阐明。图46g显示在液态电池或固态电池中,原始lgps的p和s xas峰与cv扫描至3.2v和9.8v的那些的比较。在没有机械约束的条件(表示为3.2v
‑
l)下,其中lgps和碳与粘合剂混合并在液态电池中测试,p和s两者都显示朝向高能量的明显峰位移和形状变化,指示液态单元中lgps中的显著整体氧化反应和局部原子环境的重排。而在固态电池中p峰和s峰没有显示任何整体氧化的迹象,因为没有观察到峰位移。然而,值得注意的是,在p和s光谱中肩部强度分别在2470ev和2149ev处增加。在lgps中p xas的从头多散射模拟显示在46h中,其中对单元电池施加了各种应变。实验和模拟之间的比较提示,在此xas中肩部强度的增加可能是由负应变(即,cv扫描并保持在高电压下之后结晶lgps经历的压缩)引起。如果我们将xrd中的应变加宽与xas中的肩部强度增
加联系起来,并且同时考虑到在直至10v的cv测试中没有观察到明显的分解电流,则出现与在适当机械压缩下的小的局部分解相关的物理图景。在150mpa左右的恒定外部压力且在lgps球粒中几乎为零的孔隙率下,lgps的宏观电压分解在超过电压稳定窗口(即4.0v)时在动力学上被大大抑制,没有给出li
+
离子和电子的整体转移,并且因此在cv测试中没有分解电流。然而,在lgps颗粒内部和之间仍能形成小的局部分解。由于lgps中的分解具有正反应应变,所以在机械压缩性环境下,这种小的局部分解将对邻近的结晶lgps施加压缩,诱导xrd中观察到的应变加宽和xas中观察到的肩部强度增加。xrd和xas两者都是非原位测量的事实支持我们在材料层面上的图景,即这种局部分解诱导的局部应变一旦形成,即使在电池单元层面上的外部压力已经被去除之后,也不会由于动力学障碍而被容易地释放。也就是说,适当的机械条件可以导致在lgps中从4.0v到10v的机械诱导的亚稳性,而在cv测试中没有明显的分解电流。我们的结果在此提供陶瓷硫化物的电化学窗口可以通过适当施加机械约束而显著加宽的直接证据。
[0341]
理论上,考虑其中lgps分解、gibbs能变化ag
chem
<0的无约束反应,如果:
[0342]
δg
chem
+k
eff
∈
rxn
v>0
ꢀꢀꢀ
(1)
[0343]
则该反应可以通过施加具有有效本体模量k
eff
的机械约束来抑制,其中v是参考状态体积,并且∈
rxn
是无应力反应膨胀,换句话说,∈
rxn
是在不存在任何施加的应力的情况下分解后lgps的体积分数变化。等式一的有效本体模量是与如等式28中所给出的机械约束平行地增加的陶瓷硫化物的本体模量(k
材料
):
[0344][0345]
机械约束系综中的自由能的最小化允许计算扩展的电压窗口和基态分解产物。使用从头计算数据,图47a显示在0
‑
10v的电压范围内在四个水平的机械约束(k
eff
=0gpa、5gpa、10gpa、15gpa)下对lgps的这种计算的结果。图47a1显示高于hull的能量,或分解能的量值。0ev原子
‑1的高于hull的能量指示在热力学上lgps是基态产物,而升高的值指示lgps将衰减。可以看到,高于hull的能量几乎为零(对于耐热性,<50mev)的区域的电压上限从大约2.1v增加到接近4v。图47a2显示对应于自由能最小化的基态压力。压力由k
eff
∈
rxn
给出,其中∈
rxn
对应于lgps转化成使自由能最小化的产物的体积分数。在高电压极限中,在k
eff
=15gpa下,基态压力达到4gpa,很好地对应于图46h中应变的lgps的xas模拟中使用的局部应变的水平。图47a3显示基态产物的总锂比容量,这预测在任何低于15gpa的k
eff
的条件下超过5v,lgps电解质将不会提供更大锂容量,或不能进一步分解。
[0346]
在不考虑耐热性的情况下,在不同k
eff
下的整个电压范围中通过dft预测的确切分解产物示于图47b中,确切的反应等式列于表7中。这种模拟实际上在热力学上预测由电化学驱动力诱导的小的局部分解反应,如图46中所讨论的,如何在机械压缩下定量地变化。分解中的元素价态因此可以直接与xps测量比较,xps测量对在颗粒表面上的化学价信息敏感(图47c、图47d),提供对本体敏感的xas的补充信息。化学计量的lgps由价态li
1+
、ge
4+
、p
5+
、s2‑
组成。当lgps在高电压下形成锂金属(li
1+
→
li0)时,剩余的元素必定被氧化。对于k
eff
=0gpa,我们在图47b中的模拟提示硫最可能被氧化,在2.3v之上形成s
41
‑
(lis4),并且在3.76v之上形成s0(元素硫)。根据bader电荷的dft模拟,s
41
‑
或s显示非常相似的电荷状态,并且在lgps中明显高于s2‑
,这与cv扫描至3.2v并保持10小时之后在xps中对于液态单元中的lgps观察到的大量的经氧化s一致(图47c2)。相似地,观察到在相同3.2v液态单元中p的
氧化形成ps
43
‑
中的p
5+
(图47d2)。这提示热力学有利的分解实际上代表在k
eff
=0的液态单元中实验上发生的分解(与机械压缩下的替代性动力学有利的分解相反)。
[0347]
相反,lgps的计算的热力学稳定极限在k
eff
=15gpa下达到接近4v。因此,在图47c3和图47d3中,在3.2v下在强约束的lgps的条件下没有观察到s的氧化并且观察到非常少量的氧化的p。这种少量的氧化的p可以归因于来自装置的无效约束或者电压接近热力学电压。此外,9.8v情况下,超出电压稳定极限,固态电池显示比预期少的氧化的s或p。注意,根据图47b,假定在li7ps6或li2ps3中lgps分解为s元素和氧化的p。然而,这种热力学途径被绕过。超出这种热力学稳定性,存在在高机械约束下稳定硫化物电解质的动力学因素。
[0348]
机械约束的施加可以大大降低陶瓷硫化物衰减的速度,如图53中所描绘。在衰减速率充分减慢时,有效稳定性(“机械诱导的动力学稳定性”)足够高以允许电池工作。例如,如果电解质在每个充电循环中仅衰减百万分之一,则对于仅需要持续数千次循环的实际电池设计而言,它是充分稳定的。
[0349]
提出的机械诱导的动力学稳定性的机制描绘在图53中。在给定的经历分解的lgps颗粒内,颗粒可以被划分成三个区域。前两个区域是分解的区域和原始区域,其在图53(顶部)中由分解的lgps的摩尔分数(对于纯粹分解的lgps,x
d
=1;对于原始lgps,x
d
=0)指示。第三区域是摩尔分数从0过渡到1的界面。分解前沿的传播方向由等式1的热力学关系控制。如果满足等式1,则前沿将向内传播,原始lgps优先。因此,lgps将不会分解。当违反等式1时,前沿将传播到lgps中并最终消耗颗粒。
[0350]
然而,即使当违反等式1时,前沿传播到原始lgps中的速度仍将受到机械约束的施加的影响。这在图53(底部)中示出。随着分解前沿的传播,必定存在与前沿的曲率相切的离子电流。这需要存在过电压以适应每个元素物种的前沿的有限电导率。过电压的欧姆部分由等式3的总和给出,其中ρ
i
(p)是在前沿处存在的压力(p)下每个物种i在前沿的电阻率,l
t
是分解形态的特征性长度标度,并且j
i
是离子电流密度。
[0351][0352]
考虑到ρ
i
(p)可以随着压缩而快速增长,将预期这种过电压在高压下变得显著。通过将预期的压缩与压缩单元的先前分子动力学结果进行比较,可以看出这种效应。分解前沿上的压力由p=k
eff
∈
rxn
给出,并且在该压力下材料的弹性体积应变是p=k
材料
ε
v
。由于单晶格向量的应变近似地是所以预期前沿附近的lgps的ab
‑
平面的应变在的数量级。对于其中k
eff
≈k
材料
的充分约束的系统,这种应变可以容易地达到4%,因为在高电压下∈
rxn
超过30%。考虑到预测lgps中li迁移的活化能在压缩4%时从230mev增加到590mev,则锂重新排序可以发生的速率以如下倍数降低:
[0353][0354]
可能的重新排序速率的这种量值降低的众多数量级可以解释,对于低于10v的任何电压,为什么等容单元实际上没有显示分解电流。
[0355]
图48显示使用lco、lnmo和lcmo作为阴极、lgps作为隔膜和lto作为阳极的全固态电池的恒电流循环以及其循环性能。在加压单元中进行电池测试,其中单元最初用6t压制,然后紧固在螺栓连接的[准]等容单元中。应注意,lco是包括在市售锂离子电池中的最常见和广泛使用的阴极材料,在相对于li
+
/li约4v处具有平台,而lnmo被认为是最有前景的高电压阴极材料之一,其在相对于li
+
/li的4.7v处具有平坦工作电压。lco全电池的高倍率测试示于图55中。lco和lnmo的充电和放电曲线分别描绘在图48a1和图48b1中。在第一次放电循环中,两种电池都显示出平坦的工作平台,对于lco,中心在2v(相对于li
+
/li为3.5v),而对于lnmo,中心在2.9v(相对于li
+
/li为4.4v)。此外,它们两者都表现出优异的循环性能,如图48a2和图48b2中可以观察到的,其中对于lco,在最初360次循环中容量衰退仅为9%,并且对于lnmo,在最初100次循环中容量衰退为18%。这指示阴极材料与lgps的分解或界面反应不是非常严重。这些结果与图46中报道的cv测试良好一致,其中表明机械约束可以抑制lgps的分解并且将其工作电压范围加宽至比先前报道的那些高得多的值。此外,为了进一步探测lgps的稳定性,选择先前合成的lcmo作为阴极,因为它呈现甚至比lnmo更高的工作平台。图48a3描绘lcmo相对于lto的电池测试曲线。在充电分布和放电分布两者中,可以观察到两个平台,在第一次循环的放电曲线中其中心在大约2.2v和3.2v(相对于li
+
/li为3.7v和4.7v),其分别与mn
3+
/mn
4+
和co
3+
/co
4+
的氧化反应相关。如图48b3中所示,在循环时观察到一定的容量衰退,这可能归因于在高电压状态下lcmo和lgps之间的副反应,并且对应于第50次循环中衰退33%。因此,与声称lgps的稳定窗口限于低电压范围的先前报道的结果相反,在此我们表明lgps可以用作高电压阴极全固态电池中的电解质材料,即使当充电平台高达3.8v(相对于li
+
/li为5.3v)时也显示相对良好的循环性能。图48c1
‑
48d3显示在使用lco、lnmo和lcmo作为阴极的电池循环之前和之后,xps测量的在lgps中的电子结合能。每种元素可以通过与阴极材料的化学反应(化学氧化)或通过施加电压导致的lgps的脱锂(电化学氧化)而被氧化。如图48c1
‑
48d3中所描绘,在循环之后,在硫结合电子的特征性区域中的那些电子显示出向更高能态的峰位移,指示硫已经被电化学氧化。原始样品中氧化的硫的存在指示与阴极材料的化学反应的程度。
[0356]
考虑到s,而不是p,与过渡金属(无论是来自涂覆材料还是阴极材料)结合,xas测量显示s元素的强度的预边缘,而没有发现来自p的预边缘(图48e和图56)。虽然界面反应被机械约束所规避,但是仍然存在一定量的由阴极材料和lgps之间的直接接触所发生的副反应。在电池循环之后发生更多的界面反应。
[0357]
两种材料(即,lgps和阴极材料)之间的界面反应呈现计算上的挑战,因为界面的从头模拟呈现独特的负担。取而代之地,模拟界面的化学稳定性和电化学稳定性二者的优选方法是所谓的赝相(也称为伪二进制)方法。在这些方法中,目标材料的线性组合被采用并表示为单相,其中组成和能量两者都由线性组合给出。这个相是赝相。然后,可以将常规的稳定性计算应用于赝相,以估计界面的反应能。图49a
‑
d和表6给出lgps+lno、lco、lnmo和lcmo的化学反应赝相计算的结果。在图49a
‑
d中,阴极材料(或lno)的原子分数从0扫掠到1(表示纯lgps至纯阴极或lno)。无论原子分数的哪个值使得反应能最负,都表示最坏情况的反应,并且被称为x
m
。表6给出每个界面的这些x
m
值,以及最坏情况的反应能、分解产物和表示分解界面的另外赝相。表示分解界面的这种赝相(也称为中间相)可以用来计算分解界面在电池循环时将如何进一步衰减。图49e
‑
g显示lgps+lno中间相的电化学稳定性。注意,在
阴极膜组装期间,材料一接触,lgps和阴极材料之间的化学反应就会发生。这与电化学反应相反,所述电化学反应直到外部电路组装件被附接时才发生。因此,两者之间的主要区别在于化学反应在加压/单元组装之前发生,而电化学反应在之后发生。由于化学反应在不存在完全组装的单元的情况下发生,所以初始反应总是在k
eff
=0下发生(电化学反应在完成的组装件的k
eff
下发生)。
[0358]
表6.lgps与lno、lco、lcmo或lnmo之间的界面的化学反应数据。e
rxn
是这两个相之间的最坏情况的反应能,并且x
m
是在这种最坏情况下消耗的非lgps相的原子分数。
‘
产物’列出了由这种最坏情况反应产生的相。
‘
化学分解赝相’是赝相理论在
‘
产物’中的产物集的应用。其表示一种人工相,具有它的组成相的组成、能量和体积的线性组合。
[0359][0360]
图49b
‑
d显示lco、lnmo和lcmo的化学反应能分别为345mev原子
‑1、322mev原子
‑1和335mev原子
‑1。尽管用具有124mev原子
‑1的低得多的反应能的lno(图49a)涂覆,但涂层并不完美,允许与lgps的一些接触,这种接触导致在图48c
‑
48e的原始样品中看到硫的化学氧化。图49e
‑
g显示由lgps和lno的化学反应产生的产物(构成lgps
‑
lno中间相)也经历机械诱导的亚稳性。因此,在阴极颗粒涂覆有lno的全电池中,适当的压缩(如图48中所描绘的那些电池)将导致在固态电解质的本体内以及在与阴极材料的界面处机械诱导的亚稳性。作为一般规则,相较于lgps与导体(如lco、lnmo和lcmo)的界面,lgps与绝缘体(如lno)的界面更可能经历机械诱导的亚稳性。其原因是,当中间相氧化形成锂金属时,如果界面在两种电子绝缘材料之间,则锂金属将局部地形成。然而,如果这两个相中的一个导电,则锂离子可以迁移到阳极,并且因此形成非局部相。在后一种情况下,由于形成的锂相的体积将不包括在局部体积变化中,所以局部反应膨胀将大大降低。相反,如果锂金属相局部形成,则其带来更大的局部体积变化,并且因此带来更大的反应膨胀。为此,需要以绝缘体(如lno)涂覆阴极材料用来约束,以导致在lgps的界面上的机械诱导的亚稳性。
[0361]
通常,锂金属是软的,并且由于锂即时通过本体固态电解质而导致难以施加压力。为了探测锂金属固态电池系统中加压lgps的高电压性能,使用锂金属作为阳极,石墨层作为保护层,这允许在电池测试期间施加高压。首先,如图57中所示,使用swagelok、铝加压单元和不锈钢加压单元在不同的机械条件下制造锂金属
‑
lco电池。同样,最强约束条件下的界面反应和分解反应是最低的。应用相似的结构以制造使用lcmo作为阴极的较高电压锂金属电池,其中单元初始用6t压制。图58中显示石墨保护层减轻锂金属与lgps之间的界面反应。如图59中所示,lgps本身的分解在强机械约束条件下非常小,其贡献非常小的分解电
流,如图59中所示。如图50a中所描绘,lcmo阴极然后可以充电至9v,这模拟尚未发现的高电压氧化还原化学的高电压充电状态。通过在6v、7v、8v、9v下充电lcmo,分别获得99mah/g、120mah/g、146mah/g、111mah/g的放电容量(图50a)。这指示该额外的锂容量来自lcmo的较高电压状态。尽管在电池充电至高于8v的电压后存在更多的副反应,但可以看出电池即使在高达9v时仍维持循环性能。这种高电压循环证明对于受约束的lgps的高电化学窗口超过9v。在高度脱锂状态下,阴极材料通常显示差的电化学稳定性,并且阴极材料与电解质之间的反应也更严重。
[0362]
为了将这种性能与常规电解质进行对比,图50b描绘在接近5v时失效的有机液态电解质。然而,在等容条件下测试的固态电池可以充电到9v(图50a)而没有分解平台的证据。此外,与液态电池(图50b)形成对比,在5.5v下循环且在等容条件(初始用6t压制)下测试的电池(图50c)显示即使在5.5v的高截止电压下也具有稳定的循环性能和高库仑效率。尽管由于锂金属的机械柔软性,锂金属
‑
lcmo电池的性能不如全电池好,但这种结果仍然表明,与液态电解质不同,固态电解质是运行高电压阴极材料的更好平台。
[0363]
总之,我们证明机械约束如何加宽陶瓷固态电解质的稳定性,将其电化学窗口推高到超过有机液态电解质的水平。cv测试表明,在等容条件下工作的适当设计的固态电解质可以在高达接近10v的电压下工作,而没有明显的分解迹象。提出了硫化物固态电解质的这种机械诱导的动力学稳定性的机制。此外,基于这种理解,已经表明使用最常用和最有前途的阴极材料中的一些的若干高电压固态电池单元如何能够在等容条件下工作直至9v。因此,高电压固态单元的开发不再受电解质的稳定性的折衷。我们预期这项工作是开发聚焦于非常高电压(>6v)电化学的新的能量储存系统和阴极材料的重要突破。
[0364]
方法
[0365]
样品表征
[0366]
结构分析
[0367]
在45kv和40ma下工作的rigaku miniflex 6g衍射仪中使用cukα辐射(波长为)收集常规xrd数据。工作条件是在10
°
至80
°
之间2θ扫描,步长为0.02
°
且每步的扫描速度为0.24秒。
[0368]
电化学表征
[0369]
单元的lgps+c/lgps部分是通过分别在1t、3t、6t下压制粉末而制成的球粒,并将其放入swagelok或自制的加压单元中。在cv测试中,电压从开路电压开始斜升到10v,在此期间测量在每种电压下的分解电流。cv测试在solartron 1400电化学测试系统上分别在ocv至3.2v、7.5v和9.8v之间进行,扫描速率为0.1mv/s。cv扫描后保持电压10小时以确保完全发展分解,并且在任何其他表征之前将其扫描回2.5v。在3mhz至0.1hz的范围内,在相同的机器上进行电化学阻抗谱(eis)。
[0370]
对于全固态电池,通过干法制造电极层和电解质层,所述方法采用聚四氟乙烯(ptfe)作为粘合剂,并允许获得典型厚度为100
‑
200μm的膜。另外,使用li4ti5o
12
(lto)或锂(li)金属作为阳极,组装两种不同种类的全固态电池。在任何情况下,通过混合以70:30的重量比的活性材料(lico
0.5
mn
1.5
o4、lini
0.5
mn
1.5
o4或licoo2)和li
10
gep2s
12
(lgps)粉末以及3%额外的ptfe来制备复合阴极。然后将这种混合物滚压成薄膜。一方面,对于使用lto作为阳极的那些全固态电池,采用重量比为95∶5的lgps和ptfe膜的隔膜。阳极组合物由以60∶30
∶10的重量比的lgps、lto和炭黑和3%的额外ptfe的混合物组成。最后,在填充氩气的手套箱中随后组装阴极膜(使用lico
0.5
mn
1.5
o4、lini
0.5
mn
1.5
o4或licoo2作为活性材料)/lgps膜/lto膜的swagelok电池单元。基于阳极膜中lto的量(30重量%)计算比容量。在室温下在arbinbt2000工作站上进行恒电流电池循环测试。另一方面,当使用锂金属作为阳极时,将直径和厚度分别为1/2”和40μm的li金属箔连接到集电器。为了防止界面副反应,li箔用炭黑和ptfe的重量比为96∶4的直径5/32”的炭黑膜覆盖。在将负电极装载到swagelok电池单元中之后,添加70mg充当隔膜的纯lgps粉末,并轻微压制。最后,将~1mg的阴极复合材料lcmo膜插入并压制至6tn(0.46gpa)以形成电池,其最终配置为lcmo/lgps球粒/石墨膜+li金属。对于图50a中的高电压测试,电池被充电到0.3c,随后休止30分钟,并在0.1c下放电。图50中的所有电池都是在55℃的高温下测试的。
[0371]
计算模拟
[0372]
所有从头计算和相数据都是以vienna ab
‑
initio软件包(vasp)遵循材料项目计算指南获得的。遵循在small1901470,1
‑
14(2019)和j.mater.chem.a(2019).doi:10.1039/c9ta05248h)中概述的lagrangian优化方法进行机械诱导的亚稳性计算。遵循j.mater.chem.a4,3253
‑
3266(2016),chem.mater.28,266
‑
273(2016)和chem.mater.29,7475
‑
7482(2017)的方法进行赝相计算。
[0373]
其他实施方案在权利要求书中描述。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1