含聚乙二醇化门冬酰胺酶的药物组合物及其制备方法与流程
本发明属于药物制剂
技术领域:
,尤其涉及一种注射用聚乙二醇化门冬酰胺酶的药物组合物及其制备方法。
背景技术:
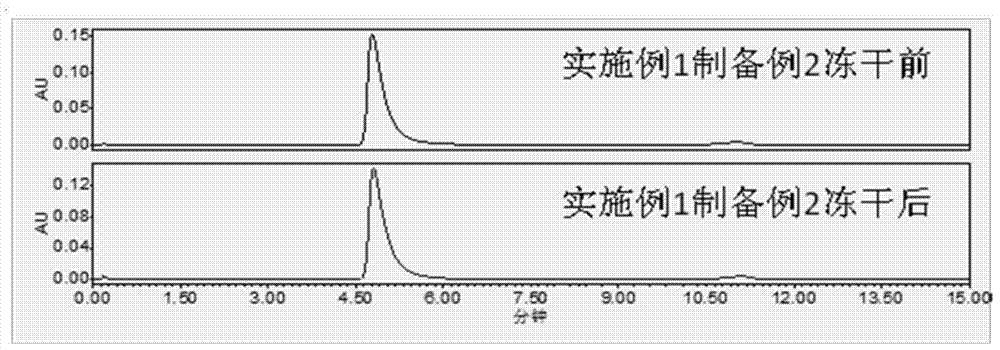
:门冬酰胺酶(Asparaginase,ASP)又名L-天冬酰胺酶或L-天门冬酰胺酶,是一种催化天冬酰胺水解成天冬氨酸的酶。门冬酰胺酶可以有效的治疗儿童或成人中的急性淋巴细胞白血病(ALL)。最近几年,含有门冬酰胺酶的药物已经用于联合化疗方案来治疗NK/T细胞淋巴瘤,并取得了较好的治疗效果。NK/T细胞淋巴瘤是中特殊类型的非霍奇金淋巴瘤,多见于亚洲和拉丁美洲,我国发病率相对较高。根据肿瘤发生部门,NK/T细胞淋巴瘤可分为鼻型NK/T细胞淋巴瘤和非鼻型NK/T细胞淋巴瘤。另外门冬酰胺酶还被用于治疗何杰金氏病,急性骨髓白血病,急性骨髓单核细胞白血病,慢性淋巴细胞白血病,淋巴肉瘤,网状细胞肉瘤和黑素肉瘤。但是,由于它来源于外源生物,对人而言是一种外源蛋白,有较强的免疫原性,临床上常见进行性免疫反应和全身性过敏反应,而限制了其临床应用。对门冬酰胺酶进行聚乙二醇修饰可以解决这个问题。国外利用聚乙二醇修饰的L-门冬酰胺酶的产品Oncaspar(Enzoninc.)于1994年上市,在2006年起被批准作为儿童和成人的ALL的一线治疗用药。国内SFDA于2009年批准恒瑞培门冬酶(聚乙二醇化门冬酰胺酶)注射液上市。国内外上市的聚乙二醇化门冬酰胺酶的药物剂型均是液体注射剂,液体注射剂有时会有被冷冻的意外,而冷冻后复融会出现聚乙二醇化门冬酰胺酶降解、活性降低的情况。此外这些液体制剂需要在2-8℃条件下保存,有效期为18个月,它们存在以下缺点:一、液体的贮存条件要求高,需要专门的冷库存放。二、运输成本高,液体注射剂在运输的过程中需要同样的温度条件,因此需采用冷链运输。三、质量风险大,贮存和运输过程中温度的意外波动,都存在导致产品质量变化的风险。四、有效期短,不利于生产销售。因此,本发明进一步希望把这种药物开发成为冻干粉针,避免液体注射剂的缺点,达到延长有效期,降低运输成本,同时降低聚乙二醇化门冬酰胺酶因贮存或运输过程中,温度条件的波动而带来的质量风险的目的。目前聚乙二醇化门冬酰胺酶的冻干粉针未见相关的研究报道。而发明人也曾尝试用已有的门冬酰胺酶冻干粉制剂配方进行聚乙二醇化门冬酰胺酶的冻干粉制剂制备,却发现有明显的降解和活性降低的情况。技术实现要素:鉴于现有产品和技术的不足,本发明的第一个目的在于提供一种冷冻后不易发生样品降解的用于肌肉或静脉注射的聚乙二醇化门冬酰胺酶制剂。技术方案如下:含聚乙二醇化门冬酰胺酶的药物组合物,含有聚乙二醇化门冬酰胺酶、缓冲剂,还含有山梨醇,所述缓冲剂选自磷酸盐或者Tris-HCl。优选地,所述药物组合物还含有保护剂和表面活性剂,保护剂选自甘露醇、蔗糖或乳糖。优选地,该聚乙二醇化门冬酰胺药物组合物的剂型为冻干粉针或者水针。优选地,上述药物组合物中聚乙二醇化门冬酰胺酶质量百分含量为5%-60%,山梨醇的质量百分含量为20%-81%,保护剂的质量百分含量为0-60%,缓冲剂是磷酸盐时,磷酸盐的质量百分含量为8%-27.9%;缓冲剂是Tris-HCl时,Tris-HCl的质量百分含量为10%-25%,表面活性剂的质量百分含量为0%-2%。优选地,上述药物组合物中保护剂和表面活性剂的含量均为0,聚乙二醇化门冬酰胺酶质量百分含量为8%-20.6%,山梨醇质量百分含量为51.5%-81%,缓冲剂的质量百分比为11%-27.9%。优选地,上述药物组合物中保护剂含量为0,聚乙二醇化门冬酰胺酶质量百分含量为18%-60%,山梨醇质量百分含量为31.5%-60%,缓冲剂的质量百分含量为8%-20%,表面活性剂的质量百分含量为0.5%-2%。优选地,上述药物组合物中表面活性剂的含量为0,聚乙二醇化门冬酰胺酶质量百分含量为5%-20%,山梨醇质量百分含量为20%-30%,缓冲剂的质量百分含量为10%-25%,保护剂含量为40%-60%。在本发明一个最优选的具体实施例中,所述的注射用聚乙二醇化门冬酰胺酶各组分的质量百分含量为:聚乙二醇化门冬酰胺酶8.0%,磷酸氢二钠9.1%、磷酸二氢钠1.9%、山梨醇81%。作为优选,所述聚乙二醇化的门冬酰胺酶是聚乙二醇随机修饰的门冬酰胺酶,其中一分子门冬酰胺酶与13-45分子聚乙二醇偶联,所述聚乙二醇为平均分子量2-20kDa的直链聚乙二醇。作为优选,PEG通过与门冬酰胺酶的游离氨基形成酰胺键或脲烷键共价连接。所述游离氨基包括赖氨酸残基和/或N-末端氨基。优选的,PEG偶联了门冬酰胺酶以后的结构通式如下:其中ASP是门冬酰胺酶,PEG是聚乙二醇部分,m是13-45的整数;n是0到3的整数。本发明的第二个目的在于提供一种注射用聚乙二醇化门冬酰胺酶冻干粉制剂的制备方法,该方法包括如下步骤:1、溶液配制:配制含山梨醇的缓冲液,降温至25℃以下,将聚乙二醇化门冬酰胺酶用上述配置好的缓冲液稀释,检测中间体。药液经0.2um微孔滤器除菌过滤,灌装,半压塞,进冻干箱。2、冷冻干燥工艺:预冻阶段:预冻采用板层全速降温,制品温度降至-45~-60℃,保持3小时,使样品完全冻结。升华阶段:开启真空泵,预抽真空至0.40mbar,开始升华,对隔板以每小时升温15-20℃的速度加热,控制样品温度接近但不超过共晶点,抽真空至0.01mbar,维持至样品水迹线消失,继续维持约2-4小时。解析干燥:对隔板以每小时升温15-20℃的速度加热,升高板层温度至20℃,当制品温度达到20±5℃以后,停止真空控制,保温干燥2-5小时,达到干燥终点,结束整个干燥过程。本申请注射用聚乙二醇化门冬酰胺酶制剂为肌肉注射型,给药剂量为2500IU/m2,每14天给药一次。与现有技术相比,本发明涉及的注射用聚乙二醇化门冬酰胺酶制剂具有如下优点:(1)稳定性高,有效期延长。本发明人尝试运用甘露醇、乳糖等常用冻干赋形剂进行冷冻干燥,但冷冻过程中聚乙二醇化门冬酰胺酶降解严重。本发明创造性地在处方中加入山梨醇,对聚乙二醇化门冬酰胺酶的活性有着特殊的保护作用。从而解决了聚乙二醇化门冬酰胺酶不能冷冻的问题,制成的冻干制剂有很好的稳定性。(2)运输方便由于聚乙二醇化门冬酰胺酶对温度敏感,运输过程中须保持2-8℃,存在着运输成本高,质量风险大的缺点。此发明的聚乙二醇化门冬酰胺酶冻干粉针在高温55℃条件下放置2个月后,酶活性无显著变化。所以本发明的聚乙二醇化门冬酰胺酶组合物在常温条件下可进行运输,既节省了运输成本,又降低了质量风险。(备注:制剂质量的“显著变化”定义为:含量与初始值相差5%;或用生物或免疫法测定时效价不符合规定。(3)冻融后不会降解原研药经过冷冻后会发生样品的降解,造成样品的失活。本发明中的液体制剂经过冷冻后,样品不发生降解,活性不会降低。(4)本发明使用的聚乙二醇化门冬酰胺酶相比市场上销售的同类产品,结构更加稳定、PEG结合牢固、不易脱落。附图说明图1注射用聚乙二醇化门冬酰胺酶冷冻干燥前后HPLC-SEC色谱图从图中可以看出,实施例1中制备例的样品在冻干前后,样品的峰形基本没有变化,说明聚乙二醇化门冬酰胺酶纯度基本不变。而实施例2中的制备例的样品,都出现了不同程度的降解。图2aHPLC-SEC分析实施例1中制备例1的样品经过冻融后的样品的峰形变化从图中可以看出,样品经过冻融后,峰形没有发生改变,说明样品中添加的辅料对样品有显著的保护作用。图2bHPLC-SEC分析培门冬酶样品经过冻融后的样品的峰形变化从图中可以看出,培门冬酶经过冻融后,出现了明显的降解峰,说明样品在冻融过程中,产生了降解产物。图3:PEG-ASP偶联物的HPLC-SEC纯度分析通过高效凝胶过滤分析结果可以看出制备的偶联物中没有明显杂质峰,纯度均大于98%。SPA5K-ASP的分子量和Oncaspar的分子量基本一致,但高于培门冬酶。由于培门冬酶也是用分子量为5K的PEG进行随机修饰的,因此说明SPA5K-ASP的修饰度明显高于培门冬酶。图4:SPA5K-ASP偶联物的SDS-PAGE电泳检测PEG修饰ASP后的蛋白电泳图。第1-3泳道的样品分别是蛋白Marker、Oncaspar、SPA5K-ASP、培门冬酶。通过蛋白电泳结果可以看出SPA5K-ASP的修饰均一性略优于培门冬酶,SPA5K-ASP的分子量高于培门冬酶。图5:Oncaspar、SPA5K-ASP和培门冬酶热处理后不同时间段的蛋白电泳考染(a)与碘染(b)结果图图5a为蛋白电泳考染(a)结果,第1-19泳道的样品分别是蛋白Marker、Oncaspar0h、Oncaspar1h、Oncaspar2h、Oncaspar3h、Oncaspar4h、Oncaspar5h、SPA5K-ASP0h、SPA5K-ASP1h、SPA5K-ASP2h、SPA5K-ASP3h、SPA5K-ASP4h、SPA5K-ASP5h、培门冬酶0h、培门冬酶1h、培门冬酶2h、培门冬酶3h、培门冬酶4h、培门冬酶5h。图5b为蛋白电泳碘染(b)结果,第1-20泳道的样品分别是蛋白Marker、SPA5KPEG、Oncaspar0h、Oncaspar1h、Oncaspar2h、Oncaspar3h、Oncaspar4h、Oncaspar5h、SPA5K-ASP0h、SPA5K-ASP1h、SPA5K-ASP2h、SPA5K-ASP3h、SPA5K-ASP4h、SPA5K-ASP5h、培门冬酶0h、培门冬酶1h、培门冬酶2h、培门冬酶3h、培门冬酶4h、培门冬酶5h。通过蛋白电泳的碘染和考染结果可以看出,SPA5K-ASP的PEG没有发生脱落,而Oncaspar和培门冬酶的PEG很容易脱落,这说明SPA-5K-ASP比Oncaspar和培门冬酶的稳定性好。图5c活性测定结果表明,在高温55℃条件下,培门冬酶在1h内基本完全失活,Oncaspar保留了30%左右的活性,而SPA5K-ASP还保留了50%左右的活性,说明其稳定性最好。图6:不同PEG-ASP偶联物荧光光谱分析使用荧光分光光度计对门冬酰胺酶、Oncaspar、SPA5K-ASP及培门冬酶进行内源荧光光谱扫描,结果表明PEG修饰后没有改变门冬酰胺酶的三级结构。图7:PEG偶联物以及培门冬酶药代动力学性质比较使用125I同位素标记示踪法对PEG修饰后的门冬酰胺酶的体内血药浓度进行了研究。从结果看,与培门冬酶相比,SPA5K-ASP的药代动力学性质略优。具体实施方式定义:本发明使用的缩写含义如下:PEG,聚乙二醇;PEG修饰剂,聚乙二醇修饰剂。聚乙二醇(PEG,HO-(CH2CH2O)n-CH2CH2OH)是一种两端带羟基的线型聚合物,聚乙二醇是经环氧乙烷聚合而成的,由重复的氧乙烯基组成,有分支型,直链型和多臂型。PEG也被称为poly(ethyleneoxide)(PEO),poly(oxy-ethylene)(POE),或者polyoxirane。一般情况下,分子量低于20,000的被称为PEG,分子量更大的被称为PEO。普通的聚乙二醇两端各有一个羟基,若一端以甲基封闭则得到甲氧基聚乙二醇(mPEG),这种衍生物是蛋白质聚乙二醇化技术中最常用到。聚乙二醇修饰剂,则是指带有官能团的聚乙二醇衍生物,是指经过活化的聚乙二醇,目前主要用于蛋白质以及多肽药物修饰,又叫修饰性聚乙二醇,修饰性PEG。M-SPA-5000,M-SPA-2000分子量分别为5000Da,2000Da的直链聚乙二醇琥珀酰亚胺丙酸酯;M-SC-10K,M-SC-5000,分子量分别为10KDa和5000Da的直链聚乙二醇琥珀酰亚胺碳酸酯;M-SCM-20KDa,分子量为20KDa的直链聚乙二醇琥珀酰亚胺乙酸酯;M-SBA-5000,分子量为5000Da的直链聚乙二醇琥珀酰亚胺丁酸酯,它们的结构通式为其中n为0时,mPEG的分子量为10KDa和5000Da时所对应的PEG类型分别是M-SC-10K,M-SC-5000;n为1时,mPEG的分子量为20KDa时所对应的PEG类型分别是M-SCM-20K;n为2时,mPEG的分子量为5000Da和2000Da时所对应的PEG类型分别是M-SPA-5000,M-SPA-2000;n为3时,mPEG的分子量为5000Da时所对应的PEG类型是M-SBA-5000;M-NPC-5000,分子量为5000Da的直链聚乙二醇硝基苯碳酸酯;结构式如下其中,mPEG的分子量为5000Da。本申请中所用,术语“偶联物”,是指聚乙二醇修饰门冬酰胺酶后得到的修饰产物;几种聚乙二醇修饰门冬酰胺酶的修饰产物可在本申请中统称为PEG-ASP或PEG修饰ASP的偶联物。本发明使用的聚乙二醇修饰剂优选下面几种:酯基活化的的聚乙二醇,更具体地,聚乙二醇修饰剂为琥珀酰亚胺丙酸酯活化的聚乙二醇。在本发明中,所修饰的门冬酰胺酶蛋白,可以是任何来源的,门冬酰胺酶可从大肠杆菌中提取,包括但不限于大肠杆菌。也可以是重组表达的。在本发明的偶联物的特定实施方式中,门冬酰胺酶与包含SEQIDNO:1的序列的蛋白具有至少约60%的序列一致性。更特别地与包含SEQIDNO:1的蛋白具有至少约65%,70%,75%,80%,85%,90%,91%,92%,93%,94%,95%,96%,97%,98%或100%的序列一致性。在特定的实施方式中,所述蛋白为大肠杆菌来源的门冬酰胺酶,其具有SEQIDNO:1的序列。SEQIDNO:1的蛋白的片段也被包含在本发明的偶联物中所使用的蛋白的定义中。所述的“SEQIDNO:1的蛋白的片段”是指多肽的序列可包括比SEQIDNO:1更少的氨基酸。本领域熟知的是,可通过置换、插入、缺失和/或添加一个或多个氨基酸来修饰多肽但同时保留其酶活性。例如,在给定的位置通过化学上等同的氨基酸来置换一个氨基酸而不影响蛋白的功能特性是常见的。因此,可预期如下改变将产生功能上等同的产物:产生以一个带负电的残基取代另一个的改变或产生一个带正电的残基取代另一个的改变。另外随着聚乙二醇合成技术的发展,可通过改变活化基团与聚乙二醇分子之间的连接基团来合成出新型的分支型聚乙二醇修饰剂。本发明专利中使用的分支型聚乙二醇修饰剂不局限于本发明中所描述的结构式。现通过以下实施例来进一步描述本发明的制备过程和实施效果,实施例仅用于例证的目的,不限制本发明的范围,同时本领域普通技术人员根据本发明所做的显而易见的改变和修饰也包含在本发明范围之内。实施例1注射用聚乙二醇化门冬酰胺酶处方(均为质量百分比)及其制备工艺制备例1:制备工艺:溶液配制:配制含5%山梨醇50mM的磷酸盐缓冲液,检测pH值在6.0-8.0降温至25℃以下,将聚乙二醇化门冬酰胺酶用上述配置好的缓冲液稀释,检测中间体。药液经0.2um微孔滤器除菌过滤,灌装压塞加盖,即得水针。如要制成冻干粉针,灌装后半压塞,进冻干箱,进行冷冻干燥工艺。冷冻干燥工艺:预冻阶段:预冻采用板层全速降温,制品温度降至-60℃,保持3小时,使样品完全冻结。升华阶段:开启真空泵,预抽真空至0.40mbar,开始升华,对隔板以每小时升温15℃的速度加热,控制样品温度不超过共晶点,抽真空至0.01mbar,维持至样品水迹线消失,继续维持约2小时。解析干燥:对隔板以每小时升温15℃速度加热,升高板层温度至20℃,当制品温度达到20±5℃以后,停止真空控制,保温干燥2小时,达到干燥终点,结束整个干燥过程。制备例2:制备工艺:溶液配制:配制含3%山梨醇100mM的磷酸盐缓冲液,检测pH值在6.0-8.0降温至25℃以下,将聚乙二醇化门冬酰胺酶用上述配置好的缓冲液稀释,检测中间体。药液经0.2um微孔滤器除菌过滤,灌装压塞加盖,即得水针。如要制成冻干粉针,灌装后半压塞,进冻干箱,进行冷冻干燥工艺。冷冻干燥工艺:预冻阶段:预冻采用板层全速降温,制品温度降至-60℃,保持3小时,使样品完全冻结。升华阶段:开启真空泵,预抽真空至0.40mbar,开始升华,对隔板以每小时升温20℃的速度加热,控制样品温度不超过共晶点,抽真空至0.01mbar,维持至样品水迹线消失,继续维持约4小时。解析干燥:对隔板以每小时升温20℃速度加热,升高板层温度至20℃,当制品温度达到20±5℃以后,停止真空控制,保温干燥5小时,达到干燥终点,结束整个干燥过程。制备例3:制备工艺:溶液配制:配制含5%山梨醇200mM的磷酸盐缓冲液,检测pH值在6.0-8.0降温至25℃以下,将聚乙二醇化门冬酰胺酶用上述配置好的缓冲液稀释,检测中间体。药液经0.2um微孔滤器除菌过滤,灌装压塞加盖,即得水针。如要制成冻干粉针,灌装后半压塞,进冻干箱,进行冷冻干燥工艺。冷冻干燥工艺:预冻阶段:预冻采用板层全速降温,制品温度降至-45℃,保持3小时,使样品完全冻结。升华阶段:开启真空泵,预抽真空至0.40mbar,开始升华,对隔板以每小时升温20℃的速度加热,控制样品温度不超过共晶点,抽真空至0.01mbar,维持至样品水迹线消失,继续维持约3小时。解析干燥:对隔板以每小时升温20℃速度加热,升高板层温度至20℃,当制品温度达到20±5℃以后,停止真空控制,保温干燥5小时,达到干燥终点,结束整个干燥过程。制备例4-8按处方(表1)配比配置溶液,其他溶液配置、冷冻干燥工艺同实施例1。表1制备例缓冲体系辅料1辅料2聚乙二醇化门冬酰胺酶制备例425%Tris-Hcl(PH8.0)30%山梨醇40%甘露醇5%制备例510%Tris-Hcl(PH8.0)20%山梨醇60%蔗糖10%制备例68%磷酸盐(PH7.0)31.5%山梨醇0.5%吐温8060%制备例715%磷酸盐(PH7.0)20%山梨醇45%乳糖20%制备例820%磷酸盐(PH7.0)60%山梨醇2%吐温8018%实施例2注射用聚乙二醇化门冬酰胺酶处方(均为质量百分比)及其制备工艺制备例1:聚乙二醇化门冬酰胺酶42.3%磷酸氢二钠47.4%磷酸二氢钠10.3%按照处方配比配制,其他溶液配置、冷冻干燥工艺同实施例1制备例2:按上述处方配比配置溶液,其他溶液配置、冷冻干燥工艺同实施例1。制备例3:按上述处方配比配置溶液,其他溶液配置、冷冻干燥工艺同实施例1。制备例4-8:按处方(表2)配比配置溶液,其他溶液配置、冷冻干燥工艺同实施例1。表2制备例缓冲体系辅料1辅料2聚乙二醇化门冬酰胺酶制备例48%Tris-Hcl(PH8.0)20%甘露醇67%蔗糖5%制备例55%Tris-Hcl(PH8.0)15%甘露醇70%乳糖10%制备例610%磷酸盐(PH7.0)49.5%海藻糖0.5%吐温8040%制备例715%磷酸盐(PH7.0)60%乳糖5%吐温8020%制备例830%磷酸盐(PH7.0)30%乳糖22%蔗糖18%实施例3注射用聚乙二醇化门冬酰胺酶冷冻干燥前后酶活性对比上述实施例及制备例制备的样品,采用中性硼硅玻璃管制注射剂瓶和溴化丁基复四氟乙烯膜包装。冷冻干燥前后测定样品的酶活性,比较冻干前后样品酶活性的变化。酶活性测定方法:对照溶液的制备取经105℃干燥至恒重的硫酸铵适量,精密称定,用水制成0.0015mol/L的溶液。磷酸盐缓冲液的制备取0.1mol/L磷酸氢二钠溶液适量,用0.1mol/L磷酸二氢钠溶液调节pH值至8.0。供试品溶液的制备取本品约0.1g,精密称定,用磷酸盐缓冲液制成门冬酰胺酶浓度为5IU/ml的溶液。测定法取试管3支(14cm×1.2cm),各加入用上述磷酸盐缓冲液配置的0.33%门冬酰胺溶液1.9ml,于37℃水浴中预热3分钟,分别于第一管(t0)中加入25%三氯醋酸溶液0.5ml,第2、3管(t)中各精密加入供试品溶液0.1ml,置37℃水浴中,准确反应15分钟,立即于第一管(t0)中精密加入供试品溶液0.1ml,第2、3管(t)中各加入25%三氯醋酸溶液0.5ml,摇匀,分别作为空白反应液(t0)和反应液(t)。精密量取t0、t和对照品溶液各0.5ml置试管中,各加水7.0ml及碘化汞钾溶液(取碘化汞23g、碘化钾16g,加水至100ml,临用前以20%氢氧化钠溶液等体积混合)1.0ml,混匀,另取试管一支,加水7.5ml及碘化汞钾溶液1.0ml作为空白对照管,室温放置15分钟,照紫外-可见分光光度法(中国药典2010版二部附录IVA),在450nm的波长处,分别测定吸光度A0、At和As,以At的平均值,按下式计算:式中5为反应常数;F为对照溶液浓度的校正值。效价单位定义:在上述条件下,一个门冬酰胺酶单位相当于每分钟分解门冬酰胺产生1μmol氨所需的酶量。蛋白含量测定方法:取本品约20mg,精密称定,照氮测定法(中国药典2010版二部附录ⅦM第一法)测定,即得。比活每1mg蛋白中含门冬酰胺酶活力的单位。表3冻干前后酶活性测定结果样品来源冻干前(IU)冻干后(IU)酶活性变化(%)实施例1制备例11511531.3%实施例1制备例2150138-8%实施例1制备例3153139-9.2%实施例2制备例115178-48.3%实施例2制备例2158114-27.8%实施例2制备例3154101-34.4%另外,实施例1中的制备例4-8中,冻干后的酶活和冻干前比较,活性损失都在5%-10%之间。而实施例2中的制备例4-8中,冻干后的酶活和冻干前比较,活性损失都大于35%。从表3可知,本发明实施例1制备的注射用聚乙二醇化门冬酰胺酶经冻干后,酶活性无显著变化,特别是制备例1。而实施例2的制备例2中,用甘露醇代替山梨醇,两种物质的化学结构式完全相同,仅空间构象有微小差别,但这两种辅料的聚乙二醇化门冬酰胺酶注射液稳定性有较大差异,含有甘露醇的样品酶活性显著下降;实施例2的制备例3中,用海藻糖代替山梨醇,海藻糖对蛋白、抗体等有很好的稳定性作用,是公认的良好的生物制剂稳定剂,但用于本品中,聚乙二醇化门冬酰胺酶的活性显著下降。实施例2的其他制备例中,用其它常用辅料的组合来考察冻干前后的样品酶活变化,发现样品和加入山梨醇的样品比较,酶活有较大的损失。实施例4注射用聚乙二醇化门冬酰胺酶冷冻干燥前后纯度对比上述实施例中制备例制备的样品,采用中性硼硅玻璃管制注射剂瓶和溴化丁基复四氟乙烯膜包装。对实施例1中制备例2和实施例2中制备例2,3,7的样品比较,比较冻干前后样品纯度的变化。样品用HPLC-SEC进行分析,结果分别见图1a,b,c,d所示由图可以看出,实施例1中制备例2的样品在冻干前后,其纯度没有发生变化,这和实施例3中的酶活变化情况相一致。而图1b,c,d中可以看出,冻干后的样品复溶后,都出现了不同程度的降解,这个结果也和实施例3中的酶活数据相一致。实施例1和2中其它制备例的冻干前后比较也是相类似的结果。即实施例1中制备例的样品在冻干前后,样品的峰形基本没有变化。而实施例2中的制备例的样品,都出现了不同程度的降解。实施例5和已上市同类药物的稳定性比较上述实施例制备的样品采用中性硼硅玻璃管制注射剂瓶和溴化丁基复四氟乙烯膜包装。取包装好的样品各20瓶,分别置加速试验条件(温度40℃±2℃,相对湿度75%±5%,2个月)、高温试验条件(温度55℃±2℃,相对湿度60%±10%,2个月)储存,测定样品酶活性。并和国内已上市的同类药物培门冬酶做比较。表4加速实验前后酶活性测定结果(单位:IU)样品来源0个月加速2个月酶活性变化实施例1制备例11531502%培门冬酶1500100%表5高温实验前后酶活性测定结果(单位:IU)样品来源0个月高温2个月酶活性变化实施例1制备例11551502%培门冬酶1490100%从表4、表5可知,实施案1制备例1的样品的加速和高温条件下酶活性基本没有变化,样品在加速和高温条件下2个月稳定。而培门冬酶样品2个月内就已经完全失活。其他制备例的样品在加速和高温条件下酶活性也基本没有变化。实施例6液体样品和已上市同类药物的冻融实验比较对实施例1中制备例1的样品冻干前的溶液,进行冻融试验(冻融试验:包括三次循环,每次循环是先于-10~-20℃放置2天,再在20℃放置2天,取样检测),并和培门冬酶进行比较。三次冻融循环后的样品用HPLC-SEC进行分析,结果分别见图2a,b所示。图2a中实施例1中制备例1的样品经过三次冻融循环后,样品的峰形没有发生变化。而图2b中培门冬酶的样品在第一次的冻融后,样品峰的右边出现了一个新的峰,说明培门冬酶样品经过一次冻融就产生了降解产物,第二次和第三次冻融后,右边出现的峰所占的比例明显提高,说明样品的降解更加严重。我们检测了这两个样品经过三次冻融循环后的酶活,结果见表6所示。表6聚乙二醇化门冬酰胺酶溶液冻融实验酶活性检测结果(单位:IU)从表6的数据可以看出,培门冬酶样品的活性显著降低,这和冻融过程中产生降解有关。而实施例1中制备例1的样品的活性经过3次冻融循环后的酶活没有发生显著改变。进一步说明辅料山梨醇对于PEG修饰的门冬酰胺酶样品有着显著的保护作用。实施例7:门冬酰胺酶的PEG偶联物的制备及分析本发明所用聚乙二醇化门冬酰胺酶通过如下方法制备、纯化、鉴定:制备例1,第一步:缓冲液置换把门冬酰胺酶的冻干粉溶于20mM的Tris-Hcl(pH8.0)缓冲液中,配制成蛋白浓度为5mg/ml的溶液。然后把样品通过AKTA层析系统的上样泵进行上样,样品吸入到Q离子交换柱(购自GE公司,HiTrapQHP5mL)上。上样结束后用A液平衡色谱柱,平衡5个柱体积后用B液进行一步洗脱,收集洗脱峰。(A液:20mM磷酸缓冲液(pH8.0),B液:20mM磷酸缓冲液+0.2M氯化钠(pH7.5)第二步:修饰反应及修饰产物纯化用M-SPA-5000(购自北京键凯科技有限公司)作为PEG修饰剂,将第一步所收集洗脱峰的门冬酰胺酶溶液按门冬酰胺酶:PEG修饰剂为1:50的摩尔比进行反应,在4℃下反应12h。其中PEG修饰反应中门冬酰胺酶的蛋白浓度为5mg/ml。反应结束后,检测结果表明门冬酰胺酶的修饰率达到了100%。反应结束后用离子交换色谱层析进行纯化。纯化的色谱法条件:Q离子交换柱(购自GE公司,HiTrapQHP5mL),C液:20mM的Tris-HCl(pH9.0),D液:含0.1MNaCl的20mM的磷酸缓冲液(pH8.0),流速2.5mL/min,检测波长为280nm。上样:上述修饰反应产物用0.5M的NaOH溶液调节至pH9.0,结合至Q离子交换柱。平衡:C液冲洗5个柱体积。洗脱:流动相配比为0-50%D液,洗脱体积为10个柱体积,洗脱时间为20分钟。门冬酰胺酶PEG偶联物样品的HPLC检测用Waters的HPLCBEH200(4.6×300mm)的分析柱进行分析,缓冲液为含0.1M硫酸钠的0.02M磷酸缓冲液(pH6.0),充分平衡上样后,以0.3ml/min的流速洗脱,检测波长为280nm,一个样品检测时间为15min。结果如图2所示。由图3可以看出,制备的偶联物中没有明显杂质,纯度均大于98%。另外,和原研药Oncaspar(由美国Enzon公司于1994年在美国上市,该产品在2009年转让给意大利的SigmaTau制药公司,本专利中所使用的Oncaspar来自于SigmaTau制药公司)相比,SPA5K-ASP的大小和Oncaspar基本相同。但和仿制药培门冬酶(购自江苏恒瑞医药股份有限公司)相比,本实施例制备的修饰产物SPA5K-ASP的出峰位置在培门冬酶出峰位置的左侧,说明其分子量大于培门冬酶,由于培门冬酶也是用分子量为5K的PEG进行随机修饰的,因此表明SPA5K-ASP上偶联的PEG的个数要比培门冬酶上偶联的PEG的个数多。由此可见,本发明的SPA5K-ASP以及原研药Oncaspar的修饰度要高于国内上市的培门冬酶。经过计算,本制备例中产品的总收率是80%。制备例2-7具体参数与得率如下表所示,表7未列出的步骤、参数与制备例一相同(所用离子交换层析柱都购自GE公司,所用修饰剂均购自北京键凯科技有限公司):表7实验证明,制备例2-7所制备的修饰产物相比已上市同类产品PEG与ASP结合更加牢固,不易脱落。下述实施例中所用的PEG-ASP偶联物均采用制备例一中所得的偶联物。实施例8:PEG化ASP修饰均一性的比较我们通过SDS-PAGE电泳比较PEG化ASP的修饰均一性。蛋白浓缩胶为5%,分离胶为8%。浓缩胶缓冲液为0.5MTris-HCl缓冲液(pH6.8);分离胶缓冲液为1.5mol/LTris-HCl缓冲液(pH8.8)。取10ug蛋白样品,与样品缓冲液等体积混合,在100℃下煮沸5min后上样运行,电泳结束后用考马斯亮蓝R250(购自国药集团)染色。由图4可以看出,与市场上的同类产品——培门冬酶(购自江苏恒瑞医药股份有限公司)和Oncaspar(购自SigmaTau制药公司)相比,我们的修饰产物SPA5K-ASP以及Oncaspar的电泳条带相对较窄,这说明SPA5K-ASP和原研药的修饰均一性均略好于培门冬酶。但并没有达到非常均一的程度,虽然门冬酰胺酶的4个亚基相同,每个亚基上课修饰的位点是相同的,但是由于空间位阻效应的存在,在修饰的过程中,已偶联上PEG分子的部位会对邻近的可修饰位点造成屏蔽效应,阻挡其它PEG分子的修饰。并且由于4个亚基空间排列结构上有微小的差异,造成相同的可修饰位点在不同的亚基上的反应性不同,综合这两种作用因素,用随机修饰的方法对门冬酰胺酶进行修饰,基本不能达到每个亚基偶联上相同数目的PEG分子。图4中,SPA5K-ASP的电泳条带明显高于培门冬酶,这也说明SPA5K-ASP的分子量明显高于培门冬酶,和实施例7中的结果相一致。由于培门冬酶也是用分子量为5K的PEG进行随机修饰的,因此说明本发明中的SPA5K-ASP的修饰度应该高于培门冬酶,但和Oncaspar基本一致。实施例9:PEG化ASP修饰位点数的确定精确量取实施例7中制备例1纯化得到的1mg/ml门冬酰胺酶(ASP)溶液40、80、120、200、300、400μl分别置于试管中,加pH8.0的0.02mol/L的pb溶液至1ml,然后加入pH9.16NaHCO3-Na2CO3缓冲液1ml,再加入0.1%TNBS溶液1ml,在混合振荡器上剧烈振荡。另取Oncaspar溶液、本申请的PEG-ASP偶联物(制备例1)、培门冬酶同上操作。上述样品于40℃反应1.5小时,然后分别加入10%SDS溶液1ml,再分别加入1MHCl0.5ml。于424nm处测定其OD值,绘制标准曲线。分别对Oncaspar样品、SPA5k-ASP、恒瑞培门冬酶样品进行了分析。其中,PEG化平均修饰率的计算:平均偶联的PEG个数的计算:平均偶联的PEG个数=PEG平均修饰度×可修饰位点,(用分子量为5000Da的PEG修饰时,可修饰位点为33个;用分子量为2000Da的PEG修饰时,可修饰位点为55个;用分子量为10000Da的PEG修饰时,可修饰位点为20个)。计算结果如表8所示。表8平均修饰度以及偶联的PEG个数样品平均修饰度偶联的PEG个数Oncaspar65.6%21.64SPA5K-ASP65%21.45培门冬酶57.4%18.942根据表8可以看出,SPA5K-ASP偶联的PEG个数高于培门冬酶样品,平均每个门冬酰胺酶的表面偶联了21.45个PEG分子,比培门冬酶多偶联2.5个PEG分子。这个结果进一步确定了SPA5K-ASP的分子量大于培门冬酶。而SPA5K-ASP和Oncaspar的修饰度基本相同。而实施例7的制备例2-7中偶联的PEG个数分别为13.1,45.2,8.5,20.3,22.5,21.6个。偶联的PEG个数随PEG分子量的增加而减少。相同分子量不同活化基团的PEG,偶联的PEG个数略有差别。实施例10:PEG-ASP偶联物的偶联基团的稳定性研究将培门冬酶、Oncaspar以及SPA5K-ASP分别用Tris-HCl(9.0)的缓冲液稀释到1mg/ml。放入55℃水浴锅中,分别于0h、1h、2h、3h、4h、5h之后各取出100ul,分别用于蛋白电泳分析以及碘染分析和酶活性检测。由图5a可以看出,和第2泳道的条带比较,第3泳道的条带明显向下迁移,说明样品的分子量出现下降,表明样品在55℃水浴条件下一个小时就出现了降解。第4-7泳道的条带继续向下迁移,说明样品不断降解。而在第8-13泳道中可以看出,蛋白的条带并没有出现显著迁移,说明样品SPA5K-ASP在此水浴条件下,5小时内并没有出现降解现象。而从14-19泳道可以看出,和Oncaspar相同,培门冬酶的样品在5h内,也出现了显著的降解,并且降解程度大于Oncaspar。由于PEG可以和碘离子形成复合物,因此通过碘染的方法可以检测PEG分子。在图5b的碘染图中可以看出,Oncaspar和培门冬酶的样品在5h内,出现了显著的降解,条带不断向下迁移,并且颜色变浅,说明其PEG不断脱落。在3-8以及15-20泳道的底部的颜色较深的条带是PEG条带,颜色逐渐变深,说明培门冬酶以及Oncaspar解离的PEG分子个数随着水浴处理时间的延长而增加。而在第9-14泳道的SPA5K-ASP样品中并没有出现条带降解的现象,说明并没有出现PEG分子解离。除了进行电泳分析,对样品还进行了酶活性的检测(参照2005版药典第二部第31页所述方法进行),结果见图5c。由图可以看出,经过水浴处理1小时,培门冬酶的活性基本完全损失,活性基本为零,Oncaspar样品的酶活略好于培门冬酶,在处理1小时后保留了30%左右的活性,而SPA5K-ASP样品在处理1小时后保留了70%左右的活性,在处理5小时后依然保持了30%左右的生物活性,稳定性显著优于培门冬酶和Oncaspar。虽然在图5a,b中,SPA5K-ASP样品并没有发生降解,但55℃加热处理,应该对门冬酰胺酶的三级结构造成了破坏,从而引起了活性降低。分析培门冬酶和Oncaspar活性显著降低的原因,应该和其PEG容易脱落有关,由于PEG分子的不断脱落,使得PEG对门冬酰胺酶的保护作用显著降低,造成活性损失较大。而SPA5K-ASP样品在处理过程中没有发生PEG脱落,PEG分子可以很好的增加门冬酰胺酶的热稳定性,因此活性损失相对较慢。同时,实施例7的制备例2-7中的产物通过稳定性考察,都没有出现PEG脱落的现象。实施例11:PEG-ASP偶联物和原蛋白的荧光谱图分析修饰蛋白以及未修饰蛋白的内源荧光检测的激发波长为280nm,发射波长范围为300~400nm。扫描速度为1200nm/min。激发和发射的缝隙宽度(slitwidths)均为5nm,使用0.1cm的样品池,在室温下进行检测。待测蛋白质的浓度范围是0.1~0.2mg/ml。通过内源荧光(IntrinsicFluorescence)来检测PEG修饰对门冬酰胺酶三级结构的影响。如图6所示,当激发波长为280nm时,门冬酰胺酶及其修饰产物的发射荧光峰值在315nm处。SPA5K-ASP样品和门冬酰胺酶以及已上市的原研药Oncaspar和仿制药培门冬酶,其荧光光谱基本一致,在315nm处有特征吸收峰。这表明PEG对门冬酰胺酶的修饰不影响其三级结构。样品吸收值有一定的差异,这可能跟样品的蛋白浓度的差异有一定关系。实施例12:门冬酰胺酶的PEG偶联物对不同肿瘤细胞的抑制作用比较为了评价PEG-ASP偶联物对肿瘤细胞的抑制率,并和培门冬酶以及Oncaspar进行比较,我们选择了THP-1(来源于人的单核细胞白血病细胞系)、Raji(来源于人的淋巴瘤细胞系)、L1210(来源于小鼠的白血病细胞)这三种细胞进行评价。通过MTT法检测细胞的抑制率,考察了不同给药浓度的抑制率,并最终计算出IC50值,计算结果如表9所示。表9SPA5K-PEG-ASP偶联物及培门冬酶对肿瘤细胞的IC50值肿瘤细胞OncasparSPA5K-ASP培门冬酶THP-17.5μmol/L3.05μmol/L7.9μmol/LRaji8.1μmol/L2.4μmol/L8.8μmol/LL121010.69μmol/L7.305μmol/L10.28μmol/L根据实验结果,本发明的PEG修饰后的门冬酰胺酶对上述3种肿瘤细胞均表现出良好的抗肿瘤作用,而本发明的PEG修饰后的门冬酰胺酶的抗肿瘤活性明显高于培门冬酶和Oncaspar。实施例13:PEG-ASP偶联物的药代动力学研究我们比较了本发明专利中制备的SPA5K-ASP和已上市的同类产品培门冬酶的药代动力学特点。我们分别考察了静脉注射和肌肉注射给药的药代动力学。对样品采用IODOGEN标记方法进行125I标记。并对标记后的供试品进行纯化,经SHPLC验证纯度。标记后的系列样品采用BCA蛋白测定试剂盒进行蛋白浓度测定,然后分别与各自一定量的未标记的样品混合,用1×的PBS缓冲液(10×的溶媒用注射用水稀释到1×)将样品稀释1.175mg/mL的注射液,并取配制后的药物(5μL左右),测定放射性。比活性=放射性/蛋白浓度。给药后按照一定的时间点进行取样,如果各静脉给药组最后一个取血时间点血药浓度未低于2min时间点血药浓度的1/20,那么接下来继续取血每天一次,直至其血药浓度低于2min时间点的1/20。如果各肌肉给药组最后一个取血时间点血药浓度未低于达峰血药浓度的1/10,那么接下来继续取血每天一次,直至其血药浓度低于达峰血药浓度的1/10。取血后将血样立即置于加有肝素钠(1000IU/mL,10μL)抗凝的EP管中,颠倒5~10次,4000rpm离心5分钟分离出血浆。取50μL血浆,加入等体积的20%三氯乙酸(TCA),涡旋混匀后,测定总放射性。之后4500rpm,常温离心10min,弃上清,测定沉淀部分放射性。代谢动力学参数计算:采用WinNonlin6.2进行非房室模型方法(NCA)拟合计算AUC等主要代谢动力学参数;2)浓度计算:3)数据处理:采用Excel2007对均值、标准差等数据进行统计描述。药代动力学各参数的计算结果见表10所示。从图7a,b的药代动力学曲线可以看出,培门冬酶和SPA5K-ASP样品通过静脉注射(iv)和肌肉注射(im)两种注射方式后的药代动力学曲线基本一致。相同剂量静脉注射给药后系统暴露的大小为SPA5K-ASP略大于培门冬酶;相同剂量肌肉注射给药后系统暴露的大小为SPA5K-ASP略大于培门冬酶。剂量经归一化后静脉和肌肉注射给药后系统暴露的大小均为SPA5K-ASP略大于培门冬酶。具体的药代动力学参数如下表10所示。表10PEG-ASP偶联物和门冬酰胺酶的药代动力学参数比较SPA5K-ASP静脉和肌肉注射给药后半衰期分别为55.8h和55.9h,培门冬酶静脉样品静脉和肌肉注射给药后半衰期分别为58.00h和46h。培门冬酶肌肉注射的半衰期略低于SPA5K-ASP。SPA5K-ASP样品的AUC都略大于培门冬酶,另外这两个样品的血浆清除率基本相同。综合分析,由于两种样品采用的都是随机修饰方式,并且所使用的PEG分子量相同,因此药代动力学性质差异不大。由于培门冬酶存在PEG脱落的现象,因此其体外稳定性会弱于SPA5K-ASP样品。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1