通过长效胰岛素制剂治疗糖尿病的制作方法
甘精胰岛素是31B-32B-Di-Arg人胰岛素(一种人胰岛素类似物)中A21位置的精氨酸进一步被甘氨酸取代而得到的。
WO2008/013938A2公开了一种含水药物制剂,其包含浓度为684U/mL的甘精胰岛素。
二甲双胍是用于治疗对饮食调整无响应的非胰岛素依赖型糖尿病(2型糖尿病)的双胍类降血糖剂。二甲双胍通过提高胰岛素敏感性和降低葡萄糖的肠道吸收来改善血糖控制。二甲双胍通常是口服给药的。
是含有甘精胰岛素的胰岛素制品,在单剂皮下注射后可以提供24小时基础胰岛素供应。
的血糖动力学(glucodynamic)效应与其它目前市售的胰岛素制品的不同之处在于,从皮下注射部位产生延迟的、可预测的甘精胰岛素吸收,形成平滑的24小时时间-浓度和作用概貌,而不会产生明确的峰。是为了满足对如下所述的长效胰岛素制品的医疗需要而开发的:能够作为每日单次注射给药,实现正常或接近正常的血糖控制,且基础胰岛素概貌在24小时内尽可能平滑。这种制备物可以提供全天的良好血糖控制,同时最大限度地减少产生低血糖的趋势,其它具有更明确的“峰”效应的胰岛素制备物可见到这种趋势。
相当数目的患者,特别是那些由于肥胖而具有更高胰岛素抵抗的患者,要使用大剂量来控制血糖。例如,100U的剂量需要注射1mLU100,其可能导致某些不适;每毫升U100含有100U(3.6378mg)甘精胰岛素。为了减少注射体积,已经开发了每mL含有300U甘精胰岛素的制剂。
实施例1所述的多中心、随机化、开放标记、平行分组研究的目的是比较U300甘精胰岛素和Lantus的疗效和安全性,它们均每天一次皮下(S.C.)给药,作为患有2型糖尿病的患者的基础-餐时(basal-bolus)胰岛素方案的一部分。患有2型糖尿病并且糖化血球蛋白A1c(HbA1c)的范围是7%-10%的患者在基础加餐时胰岛素方案中注射至少42U Lantus U100或相当量的中性鱼精蛋白锌(NPH)胰岛素,这样的患者对本研究是适格的。这些接受着相对高剂量基础胰岛素的患者受益于U300胰岛素制剂相比于U100制剂的较低注射体积。
每毫升甘精胰岛素U300含有300U(10.9134mg)甘精胰岛素。这种制剂使得患者能够以三分之一的注射体积注射相同单位数的甘精胰岛素。这种制剂在本文中也被称作HOE901-U300。
在实施例1所述的研究中,将患者根据其HbA1c分层(<8.0%;≥8.0%)。主要功效分析根据从基线到终点的HbA1c变化来测试甘精胰岛素U300相比于Lantus的非劣性(non-inferiority)(安排在第6个月;非劣性边际0.4%HbA1c单位)。HbA1c反映了数月内的平均血糖,并对于糖尿病并发症具有强预测价值。6个月的研究治疗持续时间被认为足以在从Lantus或NPH胰岛素换为甘精胰岛素U300之后实现稳态条件,使得HbA1c的时间依赖性变化和与之相伴的低血糖风险能够得到充分评估。
主要的次级终点包括夜间低血糖。低血糖在糖尿病的短期和长期血糖管理中是一个关键的限制因素。尽管糖尿病的血糖管理已经得到了稳步改善,但是基于群体的数据表明,低血糖仍然是1型和2型糖尿病患者的主要问题(American diabetes association,workgroup on hypoglycemia:Defining and Reporting Hypoglycemia in Diabetes.Diabetes Care 28(5),2005,1245-1249)。
在实施例1所述的研究中,令人惊讶地发现,用甘精胰岛素U300制剂治疗2型糖尿病患者,与用U100治疗相比,夜间低血糖事件的风险可以显著减少。从第9周开始到6个月之间具有至少一次夜间重度低血糖和/或确认的低血糖的患者发生率在U300组中[136/404(33.7%)]比Lantus组中[180/400(45.0%)]低(见表6)。显示出U300对Lantus的优势,其相对危险度为0.75(95%CI[0.63,0.89])(p=0.0010)。
实施例2描述的临床试验比较了甘精胰岛素U300(HOE901-U300)制剂与U 100各自在2型糖尿病患者中与口服抗高血糖药物联用时的疗效和安全性。
在实施例2中,U300相比于Lantus的非劣性被相对于Lantus的HbA1c最小二乘平均值差-0.01%(95%CI[-0.139;0.119])所证明(表26)。注射前SMPG的最小二乘平均值变化与U300(-0.56mmol/L)和Lantus组(-0.51mmol/L)相似(表28)。
实施例2确认,通过用甘精胰岛素U300制剂治疗,夜间低血糖事件比用U100治疗显著减少。在一个不同的患者群中,即用抗高血糖药物没有单独得到充分控制的2型糖尿病患者中,令人惊讶地发现,发生至少一次夜间重度低血糖和/或确认的低血糖的患者的发生率在U300组[87/403(21.6%)]中相比于Lantus组[113/405(27.9%)]要低(见表27)。U300相比于Lantus的优势由0.77的相对危险度(95%CI[0.61,0.99])(p=0.0380)所显示。
实施例3比较了每天一次给药U300甘精胰岛素制剂与餐时胰岛素组合的可调适时间间隔与固定给药间隔。实施例3是实施例1所述试验的子研究。没有观察到对HbA1c(表50)和空腹血浆葡萄糖(表51)的负面作用。两个方案中的低血糖总发生率相似,而与低血糖的分类无关(表53)。
实施例6比较了每天一次给药U300甘精胰岛素制剂与口服抗高血糖药物组合的可调适时间间隔与固定给药间隔。实施例6是实施例2所述试验的子研究。没有观察到对HbA1c(表67)和空腹血浆葡萄糖(表68)的负面作用。两个方案中的低血的总发生与相似,而与低血糖的分类无关(表70)。
本发明的一个方面涉及用于1型或2型糖尿病治疗的含水药物制剂,其中该治疗可减少夜间低血糖的风险,所述制剂含有200-1000U/mL甘精胰岛素[等摩尔于200-1000IU人胰岛素],但是所述制剂的浓度不是684U/mL甘精胰岛素。本发明涉及用于减少夜间低血糖的风险的含水药物制剂。
本发明的制剂在施用给糖尿病患者时能够减少夜间低血糖的发生率,如本文所述。“减少夜间低血糖的发生率”包括减少夜间低血糖事件的次数,和/或减少夜间低血糖事件的严重程度。本文所述的制剂适用于减少夜间低血糖的发生率。
本发明的制剂在施用给糖尿病患者时能够防止夜间低血糖,如本文所述。“防止夜间低血糖”包括减少夜间低血糖事件的次数,和/或减少夜间低血糖事件的严重程度。本文所述的制剂适用于防止夜间低血糖。
本发明的制剂适用于减少夜间低血糖事件的次数,和/或夜间低血糖事件的严重程度。
在本发明中,低血糖是这样的状况,其中2型糖尿病患者经历低于70mg/dL(或低于3.9mmol/L),低于60mg/dL(或低于3.3mmol/L),低于54mg/dL(或低于3.0mmol/L),低于50mg/dL,低于40mg/dL,或低于36mg/dL的血浆葡萄糖浓度。
在本发明中,“症状性低血糖”或“症状性低血糖事件”是与由低血糖导致的临床症状相关的状况,其中血浆葡萄糖浓度可能低于70mg/dL(或低于3.9mmol/L),低于60mg/dL(或低于3.3mmol/L),低于54mg/dL(或低于3.0mmol/L),低于50mg/dL,或低于40mg/dL。临床症状可以是,例如,出汗,心悸,饥饿,不安,焦虑,疲乏,烦躁不安,头痛,集中力缺失,嗜睡,精神障碍,视觉障碍,短暂性感官缺陷,瞬时运动缺陷,精神错乱,惊厥和昏迷。在本发明的方法中,可以选择症状性低血糖的一种或多种临床症状,如本文所指出的。症状性低血糖可能与口服糖后的迅速恢复相关。
在本发明中,“重度症状性低血糖”或“重度症状性低血糖事件”是这样的状况,其具有如本文所指出的由于低血糖导致的临床症状,其中血浆葡萄糖浓度可以低于70mg/dL(或低于3.9mmol/L),低于54mg/dL(或低于3.0mmol/L),或低于36mg/dL(或低于2.0mmol/L)。重度症状性低血糖可能伴有由于低血糖事件导致的急性神经功能缺损。在重度症状性低血糖中,患者可能需要其他人的帮助来主动给予糖、胰高血糖素、或其他复苏操作。这些症候可能伴有足以诱发癫痫、意识障碍或昏迷的神经低血糖症。在这样的事件的过程中可能无法进行血浆葡萄糖测量,但是可以认为由于血浆葡萄糖恢复正常所导致的神经恢复足以证明该事件是血浆葡萄糖浓度低所诱导的。
重度症状性低血糖的定义可包括所有这样的症候:其中神经障碍严重到足以阻碍自我治疗,并因此认为这些症候将患者置于伤害自身或他人的风险之中。急性神经损伤可以是选自下面的至少一种:嗜睡,精神障碍,视觉障碍,短暂性感官缺陷,瞬时运动缺陷,精神错乱,惊厥,和昏迷。“需要帮助”意思是指患者不能自助。在没有帮助的需要时出于善意帮助患者,不应视为“需要帮助”的事例。
重度症状性低血糖可能伴随口服糖、静脉内葡萄糖和/或胰高血糖素给药后的迅速恢复。
在本发明中,“有记录的症状性低血糖”或“有记录的症状性低血糖事件”是这样的事件:在该事件期间,典型的低血糖症状伴随测得的血浆葡萄糖浓度≤70mg/dL(≤3.9mmol/L),或小于或等于54mg/dL(≤3.0mmol/L)。被认为是由于低血糖症候导致的临床症状包括,例如:多汗、精神紧张、乏力/虚弱、震颤、头晕、食欲增加、心悸、头痛、睡眠障碍、精神错乱、抽搐、神志不清、昏迷。
在本发明中,“无症状低血糖”或“无症状低血糖事件”是这样的事件,其不伴随典型低血糖症状,但是测得的血浆葡萄糖浓度小于或等于70mg/dL(3.9mmol/L),或者小于或等于54mg/dL(3.0mmol/L)。
在本发明中,“可能发生的症状性低血糖”或“可能发生的症状性低血糖事件”是这样的事件,在事件期间,低血糖症状并不伴随血浆葡萄糖测定,但是可推定是由于血浆葡萄糖浓度小于或等于70mg/dL(或者小于或等于3.9mmol/L),或者小于或等于54mg/dL(或者小于或等于3.0mmol/L)导致的;症状用口服糖治疗,而没有测量血浆葡萄糖。
在本发明中,“相对低血糖”或“相对低血糖事件”是这样的事件,事件期间糖尿病患者报告有任何典型的低血糖症状,并且将该症状解释为低血糖的指示,但是测得的血浆葡萄糖浓度大于70mg/dL(或者大于3.9mmol/L)。
在本发明中,“夜间低血糖”或“夜间低血糖事件”是在夜间发生的属于上文所述的任何低血糖分类的低血糖事件。“夜间低血糖”可以由钟表时间定义。特别地,夜间低血糖是发生在00:00-05:59a.m.之间的低血糖。患者可以是清醒的,或者可能由于该事件而醒来。患者也可能在该事件期间是睡着的。
在本发明中,“日间低血糖”或“日间低血糖事件”具体指在06:00a.m.-23:59之间发生的属于如上所述低血糖分类的任何低血糖。
在本发明中,夜间低血糖可以是症状性低血糖、重度症状性低血糖、有记录的症状性低血糖、可能发生的症状性低血糖、相对症状性低血糖、或无症状低血糖。优选的是症状性低血糖,更优选地是重度症状性低血糖。
如本文所使用的,“减少低血糖风险”可以包括减少低血糖发生率。每患者年的低血糖发生率对每位患者可以计算为:365.25x(发生低血糖的次数)/(暴露的天数),并按照事件类型和治疗组别汇总。如本文所使用的,“减少低血糖风险”可以进一步包括当如本文所述地向糖尿病患者施用本文所述的制剂时,防止患者的低血糖。如本文所使用的,“减少低血糖风险”可进一步包括减少夜间低血糖事件的数目,和/或减少夜间低血糖事件的严重程度。
实施例3和6证明,甘精胰岛素U300注射时间的偶尔调适对HbA1c(表50和67)和空腹血浆葡萄糖(表51和69)没有负面作用。以可调适时间间隔施用和以固定给药间隔施用低血糖的总体发生率相似,且与低血糖类型无关(表53和70)。
本发明的一个方面涉及以可调适的时间间隔施用的含水药物制剂。这个方面涉及用于1型和2型糖尿病治疗的含水药物制剂,其中该制剂每天给患者施用一次,并且其中在每周中至少2天距前次施用的时间间隔范围是24.5h-28h或20h-23.5h,和其中距前次施用的平均时间间隔是大约24h,所述制剂包括200-1000U/mL甘精胰岛素[等摩尔于200-1000IU人胰岛素],但所述制剂的浓度不是684U/mL甘精胰岛素。
如本文所使用的,“距前次施用的时间间隔”是两次连续施用,特别是两次注射之间的时间间隔。
优选地,制剂包括300U/ml甘精胰岛素。
在治疗方案中,制剂可以在某个固定时间左右的一定时间范围内施用,例如在晚间或者晨间的某个固定时间左右施用。距前次施用的平均时间间隔是大约24h(见表46)。“大约24h”的间隔特别是指24h+/-10min的范围,24h+/-20min的范围,或24h+/-30min的范围。平均时间间隔可以基于,例如,每周、每月或每2或3个月进行计算,或者可以基于更长的时间计算。
表47描述了实施例3中试验组和对照组患者的给药方案顺应性。描述了不同给药间隔分类下患者进行的注射的%。在对照组中(24h的固定给药间隔),大约88%的U300剂量以距前次注射23-25h的间隔注射。大约12%的剂量以短于23h或超过25h的间隔注射。在考虑每天注射一次的情况下,患者每周少于一天以短于23h或长于25h的间隔给予U300制剂。在试验组中(可调适的时间间隔),大约63%的U300剂量以距前次注射23-25h的间隔注射。大约37%的剂量以短于23h或超过25h的间隔注射。在考虑每天注射一次的情况下,患者在每周有2或3天以短于23h或长于25h的间隔给予U300制剂。
含水制剂可以在每周至少两天,每周至少3天,每周至少4天,或者每周至少5天以本文指定的时间间隔施用。含水制剂可以在每周最多5天,每周最多4天,或者每周最多3天以本文指定的时间间隔施用。更具体地,含水制剂在每周有2或3天、或者每周有2至3天以本文指定的时间间隔施用。
“偶尔调适(Occasional adaptation)”具体意指含水制剂每周有2天或3天按照本文指定的时间间隔施用。
如本文所指出的,“每周天数”可以例如基于每周,基于每月,或者基于每2个或3个月计算,或者可以以更长的时间基础计算。
“可调适的注射间隔”意思是距前次注射的时间间隔可以在预定的时间范围内变化。距前次施用的时间间隔可以在24.5h-28h的范围内或者在20h-23.5h的范围内。具体地,距前次施用的时间间隔可以在25h-28h的范围内或者在20h-23h的范围内。
距前次施用的时间间隔也可以在25h-27h的范围内或者在21h-23h的范围内。
距前次施用的时间间隔可以在25h-26.5h的范围内或者在21.5h-23h的范围内。
在这个方面,制剂的赋形剂可以是如本文所述的赋形剂。被治疗的患者可以是如本文所述的患者。
如本文所述,可调适时间间隔的治疗方案可以与夜间低血糖风险的减少相结合,如本文所述。
如本文所述的以可调适时间间隔给药的适用于1型或2型糖尿病治疗的制剂可以与可减少夜间低血糖风险的1型或2型糖尿病治疗联用。
在本发明中,正常血糖可以指这样的血浆葡萄糖浓度:70mg/dL-140mg/dL(相应于3.9mmol/L-7.8mmol/L)。
要用本文所述制剂治疗的患者可以是1型或2型糖尿病患者。优选地,患者是2型糖尿病患者。
本发明的药物制剂可以和至少一种抗高血糖剂联合给药。特别地,该至少一种抗高血糖剂是二甲双胍或/和其药物可接受的盐。二甲双胍是1,1-二甲基双胍(CAS号657-24-9)的国际通用名。在本发明中,术语“二甲双胍”包括其任何药物可接受的盐。
在本发明中,二甲双胍可以口服给药。技术人员知道适合于口服治疗糖尿病的二甲双胍制剂。二甲双胍可以根据需要以足够诱致治疗效果的量施用给患者。二甲双胍可以以至少1.0g/天或至少1.5g/天的剂量施用。对于口服施用,二甲双胍可以配制成固体剂量形式,例如药片或药丸。二甲双胍可以用合适的药物可接受的载体、佐剂或/和辅助物质配制。
本发明的制剂和二甲双胍可以通过不同的给药途径给药。二甲双胍可以口服给药,而本发明的制剂可以肠胃外给药。
要用本发明制剂治疗的患者可以是正在罹患2型糖尿病的患者,其中2型糖尿病单独用至少一种抗高血糖剂未得到充分控制。抗高血糖剂可以是二甲双胍,其中施用未充分控制2型糖尿病,例如在治疗至少2个月或至少3个月后未充分控制,例如用例如至少1.0g/天或至少1.5g/天的二甲双胍剂量治疗至少2个月或至少3个月后未充分控制。
在本发明中,如果至少一个描述血糖浓度的生理参数(例如HbA1c值、注射前SMPG和/或空腹血浆葡萄糖浓度)超过正常血糖值,如本文所述地,则2型糖尿病患者未得到充分控制。特别地,未得到充分控制的2型糖尿病患者可能具有:
(i)HbA1c数值范围是7%-10%或者更大,
(ii)注射前SMPG至少为9mmol/L,或/和
(iv)空腹血浆葡萄糖至少为8.0mmol/L。
要用本文所述制剂治疗的患者的HbA1c值范围是7%-10%。更具体地,在治疗开始时,待治疗患者在开始本发明治疗时的HbA1c值是至少8%,或者HbA1c值的范围是8%-10%。
要用本文所述制剂治疗的患者可以是成年受试者。在本发明治疗开始时,患者的年龄可以是至少50岁,至少57岁,至少58岁,至少59岁,至少60岁,至少65岁,至少70岁,或至少75岁。
在本发明治疗开始时,要用本文所述制剂治疗的患者可以是肥胖受试者。在本发明中,肥胖受试者在治疗开始时的体重指数(BMI)可为至少30kg/m2,至少31kg/m2,至少32kg/m2,至少33kg/m2,至少34kg/m2,至少35kg/m2,至少36kg/m2,至少37kg/m2,至少38kg/m2,至少39kg/m2或至少40kg/m2。优选地,患者在治疗开始的BMI为至少34kg/m2或至少36kg/m2。
要用本文所述制剂治疗的患者可能具有增加的低血糖风险,特别是经历过至少一次低血糖事件的2型糖尿病患者。
要用本文所述制剂治疗的患者在即将进行本文所述的治疗之前可能接受了胰岛素。特别地,患者可能接受了基础胰岛素,例如剂量为至少32U/日或者至少42U/日。在本发明中,任何使用基础胰岛素的预治疗均可以考虑。特别地,基础胰岛素可以选自甘精胰岛素,地特胰岛素(Detemir),NPH,慢胰岛素(Lente),超慢胰岛素(Ultralente),诺和灵(Novolin),优泌乐(Humalog)和其混合物。混合物可以包含两种不同的基础胰岛素。例如,可以采用包含地特胰岛素和甘精胰岛素的混合物或者包含NPH和诺和灵的混合物。优选地,基础胰岛素是甘精胰岛素,或者包含甘精胰岛素的混合物。在本发明中,“基础胰岛素”包括其合适的药物可接受的盐。
要用本文所述制剂治疗的患者在即将进行本文所述的治疗之前可能接受了餐时短效胰岛素。餐时短效胰岛素可以是胰岛素类似物,例如赖谷胰岛素、赖脯胰岛素、或门冬胰岛素。
本文所述的制剂可以每天施用一次或两次。特别地,本文所述的制剂可以每天施用一次,例如在夜间。本文所述的制剂可以每天在夜间预定的时间施用一次。
患者可以额外接受餐时短效胰岛素。餐时短效胰岛素可以是胰岛素类似物,例如赖谷胰岛素、赖脯胰岛素、或门冬胰岛素。
要用本文所述制剂治疗的患者在本发明治疗开始时的注射前自我监测血浆葡萄糖(SMPG)浓度可以是至少9mmol/L,至少10mmol/L,至少10.5mmol/L,或至少11mmol/L。在本发明中,自我监测血浆葡萄糖可以是空腹SMPG或注射前SMPG(例如,在注射本文所述制剂之前30分钟测得的)。
要治疗的患者在本发明治疗开始时的空腹血浆葡萄糖浓度可为至少7mmol/L,至少7.5mmol/L,至少8mmol/L,至少8.5mmol/L或至少9mmol/L。
本发明并不仅限于甘精胰岛素U 300制剂,其它更高浓度的甘精胰岛素制剂,例如在说明书中详细列出的,也是有效的,但是本文所述的临床研究是用甘精胰岛素U 300制剂实施的。
1mL甘精胰岛素U300制剂含有10.913mg 21A-Gly-30Ba-L-Arg-30Bb-L-Arg人胰岛素[等摩尔于300IU人胰岛素],90μg锌,2.7mg间甲酚,20mg甘油85%,HCl和NaOH,调节至pH 4.0;比重1.006g/mL。
然而,赋形剂类型及其浓度的变化是可能的。
药物制剂含有200–1000U/mL甘精胰岛素[等摩尔于200-1000IU人胰岛素],其中所述制剂的浓度不是684U/mL,优选地是250-500U/mL甘精胰岛素[等摩尔于250-500IU人胰岛素],更优选地270–330U/mL甘精胰岛素[等摩尔于270–330IU人胰岛素],甚至更优选地是300U/mL甘精胰岛素[等摩尔于300IU人胰岛素]。
药物制剂可以添加表面活性剂,例如,除其他外,非离子表面活性剂。特别地,制药惯用的表面活性剂是优选的,例如:多元醇如甘油、山梨醇等的部分脂肪酸酯和脂肪酸酯以及醚类(特别是20和80,),或泊洛沙姆。药物组合物中存在的表面活性剂的浓度是5-200μg/ml,优选地5–120μg/ml,特别优选地20–75μg/ml。
制剂可以额外含有防腐剂(例如苯酚,间甲酚,对甲酚,对羟苯甲酸酯),等渗剂(例如甘露糖醇,山梨糖醇,乳糖,右旋糖,海藻糖,氯化钠,甘油),缓冲物质、盐、酸和碱,并进一步含有赋形剂。这些物质各自可以单独存在或者可选地作为混合物存在。
甘油、右旋糖、乳糖、山梨醇和甘露醇在药物制备物中的存在浓度可以是100–250mM,NaCl浓度最高为150mM。缓冲物质,例如磷酸盐、乙酸盐、柠檬酸盐、精氨酸、甘氨酰甘氨酸或TRIS(即2-氨基-2-羟甲基-1,3-丙二醇)缓冲液和相应盐的存在浓度为5–250mM,优选的10–100mM。其它赋形剂可以是,除其他外,盐或精氨酸。
制剂的锌浓度范围是通过0-1000μg/mL,优选地20–400μg/mL,最优选地90μg/mL的锌的存在所达到的浓度范围。然而,锌可以氯化锌形式存在,但是盐并不限于是氯化锌。
在药物制剂中,甘油和/或甘露醇的存在浓度可以是100–250mmol/L,和/或NaCl优选以最多150mmol/L的浓度存在。
在药物制剂中,缓冲物质可以以5–250mmol/L的浓度存在。
本发明进一步的主题是适用于本文所述用途的药物胰岛素制剂,其含有进一步的添加剂,例如可延缓胰岛素释放的盐。这样的缓释胰岛素与上述制剂的混合物包含在其中。
本发明进一步的主题涉及用于产生适用于本文所述用途的此类药物制剂的方法。为了产生这些制剂,将各成分溶解在水中,并且用HCl和/或NaOH对pH进行调节。类似地,本发明进一步的主题涉及这些制剂治疗糖尿病的用途。
本发明进一步的主题涉及在产生胰岛素、胰岛素类似物或胰岛素衍生物或其制备物的过程中使用或者添加表面活性剂作为稳定化剂。
本发明进一步涉及如上所述的制剂,其还额外包含胰高血糖素样肽-1(GLP1)或其类似物或衍生物,或者毒蜥外泌肽(exendin)-3或-4或其类似物或衍生物,优选地毒蜥外泌肽-4。
本发明进一步涉及如上所述的制剂,其中毒蜥外泌肽-4的类似物选自包含下述的组:
H-desPro36-毒蜥外泌肽-4-Lys6-NH2(利西拉来(Lixisenatide),AVE0010),
H-des(Pro36,37)-毒蜥外泌肽-4-Lys4-NH2和
H-des(Pro36,37)-毒蜥外泌肽-4-Lys5-NH2,
或其药理学可耐受的盐。
本发明进一步涉及如上所述的制剂,其中毒蜥外泌肽-4的类似物选自包含下述的组:
desPro36[Asp28]毒蜥外泌肽-4(1-39),
desPro36[IsoAsp28]毒蜥外泌肽-4(1-39),
desPro36[Met(O)14,Asp28]毒蜥外泌肽-4(1-39),
desPro36[Met(O)14,IsoAsp28]毒蜥外泌肽-4(1-39),
desPro36[Trp(O2)25,Asp28]毒蜥外泌肽-2(1-39),
desPro36[Trp(O2)25,IsoAsp28]毒蜥外泌肽-2(1-39),
desPro36[Met(O)14Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)和
desPro36[Met(O)14Trp(O2)25,IsoAsp28]毒蜥外泌肽-4(1-39),
或其药理学可耐受的盐。
本发明进一步涉及在前一段中描述的制剂,其中肽-Lys6-NH2被附接于毒蜥外泌肽-4的类似物的C端。
本发明进一步涉及如上所述的制剂,其中毒蜥外泌肽-4的类似物选自包含下述的组:
H-(Lys)6-des Pro36[Asp28]毒蜥外泌肽-4(1-39)-Lys6-NH2
des Asp28Pro36,Pro37,Pro38毒蜥外泌肽-4(1-39)-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Asp28]毒蜥外泌肽-4(1-39)-NH2,
H-Asn-(Glu)5des Pro36,Pro37,Pro38[Asp28]毒蜥外泌肽-4(1-39)-NH2,
des Pro36,Pro37,Pro38[Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-des Pro36,Pro37,Pro38[Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36[Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-Lys6-NH2,
H-des Asp28Pro36,Pro37,Pro38[Trp(O2)25]毒蜥外泌肽-4(1-39)-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-NH2,
H-Asn-(Glu)5-des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-NH2,
des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-Lys6-NH2,
des Met(O)14Asp28Pro 36,Pro37,Pro38毒蜥外泌肽-4(1-39)-NH2,
H-(Lys)6-des Pro36,Pro 37,Pro38[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-NH2,
H-Asn-(Glu)5-des Pro36,Pro37,Pro38[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-NH2,
des Pro36,Pro37,Pro38[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-Lys6-NH2,
H-Asn-(Glu)5des Pro36,Pro37,Pro38[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36[Met(O)14,Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-Lys6-NH2,
des Asp28Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25]毒蜥外泌肽-4(1-39)-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-NH2,
H-Asn-(Glu)5-des Pro36,Pro37,Pro38[Met(O)14,Asp28]毒蜥外泌肽-4(1-39)-NH2,
des Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-des Pro36,Pro37,Pro38[Met(O)14,Trp(O2)25,Asp28]毒蜥外泌肽-4(1-39)-(Lys)6-NH2,
或其药理学可耐受的盐。
本发明进一步涉及如上所述的制剂,其额外包含Arg34,Lys26(Nε(γ-谷氨酰(Nα-十六酰)))GLP-1(7-37)[利拉鲁肽]或其药理学可耐受的盐。
在一个实施方案中,本发明涉及适用于本文所述用途的含水药物制剂,其包含的甘精胰岛素的范围是200–1000U/mL[等摩尔于200-1000IU人胰岛素],优选地200U/ml-650U/mL,更优选地700U/mL-1000U/ml,更优选地270–330U/mL,最优选的浓度是300U/mL,但所述制剂的浓度不是684U/mL甘精胰岛素。
此外,制剂还包含毒蜥外泌肽-4的类似物,例如利西拉来、艾塞那肽(exenatide)和利拉鲁肽(liraglutide)。这些毒蜥外泌肽-4类似物在制剂中的存在浓度范围是0.1μg-10μg/U甘精胰岛素,优选地0.2-1μg/U甘精胰岛素,更优选地0.25μg-0.7μg/U甘精胰岛素。利西拉来是优选的。
此外,含水药物制剂包含选自下组的一种或多种赋形剂:锌、间甲酚、甘油、聚山梨醇酯20和钠。具体而言,含水药物制剂可包含90μg/mL锌,2.7mg/mL间甲酚和20mg/ml的甘油85%。任选地,含水药物制剂可包含20μg/mL聚山梨醇酯20。
本文所述的含水药物制剂的pH可以是4.6或更低,优选地4.5或更低。
本文所述的含水药物制剂的pH也可以是3.4-4.6的范围,优选地4-4.5的范围。
本发明的另一个方面涉及用于治疗如本文所述的疾病或状况的方法,特别是用于治疗1型或2型糖尿病的方法,包括向所述糖尿病患者施用本发明的含水药物组合物,其中该治疗可减少夜间低血糖的风险。该方法优选地是指2型糖尿病的治疗。在公开的各种浓度范围中,优选的是300U/mL。进一步,含水药物制剂还可以包括锌、间甲酚、甘油、聚山梨醇酯20和钠以及它们的混合物,它们处于本文公开的与本发明含水药物制剂相关的范围内。在优选的实施方案中,含水药物制剂还包括0.1μg-10μg利西拉来/U甘精胰岛素。夜间低血糖可以是任何如本文所定义的夜间低血糖。患者可以是任何如本文所定义的患者。
胰岛素优选地每天给药一次,但是可以根据需要每天给药2次。剂量要求随着各个患者为实现正常或可接受的血糖水平而确定的需要而变化。
方法还可以是治疗患者中1型或2型糖尿病的方法,包括向所述患者施用如本文所述的含水药物组合物,其中制剂每天给药一次,并且其中每周有至少2天距前次施用的时间间隔在24.5h-28h或者20h-23.5h的范围,且其中距前次施用的平均时间间隔是大约24h。时间间隔可以是如本文定义的时间间隔。含水制剂可以每周至少3天,每周至少4天,或者每周至少5天以本文指定的时间间隔施用。该方法优选地指2型糖尿病的治疗。在公开的各种浓度范围中,优选的浓度是300U/mL。进一步,含水药物制剂还可以包括锌、间甲酚、甘油、聚山梨醇酯20和钠以及它们的混合物,它们均处于本文公开的与本发明含水药物制剂相关的范围内。在优选的实施方案中,含水药物制剂还包括0.1μg-10μg利西拉来/U甘精胰岛素。患者可以是如本文所定义的任何患者。
如本文所述的以可调适的时间间隔施用的1型或2型糖尿病治疗方法可以和如本文所述的可减少夜间低血糖风险的1型或2型糖尿病治疗方法组合。
本发明的另外一个方面涉及如本文所述的含水制剂供制造用于治疗如本文所述的疾病或病症,特别是用于治疗1型或2型糖尿病的药物的用途,其中该治疗可减少夜间低血糖的风险。该用途优选地是指2型糖尿病的治疗。在公开的各种浓度范围中,优选的是300U/mL。进一步,含水药物制剂还可以包括锌、间甲酚、甘油、聚山梨醇酯20和钠以及它们的混合物,它们均处于本文公开的与本发明含水药物制剂相关的范围内。在优选的实施方案中,含水药物制剂还包括0.1μg-10μg利西拉来/U甘精胰岛素。夜间低血糖可以是如本文所定义的任何夜间低血糖。患者可以是如本文所定义的任何患者。
胰岛素优选地每天给药一次,但是可以根据需要每天给药2次。剂量要求随着各个患者为实现正常或可接受的血糖水平而确定的需要而变化。
本发明的另一个方面涉及如本文所述的含水制剂供制造用于治疗患者1型或2型糖尿病的药物的用途,所述治疗包括向患者施用如本文所述的含水药物组合物,其中制剂每天施用一次,并且其中每周至少2天距前次施用的时间间隔在24.5h-28h或者20h-23.5h的范围,并且其中距前次施用的平均时间间隔是大约24h。时间间隔可以是如本文定义的时间间隔。含水制剂可以每周至少3天,每周至少4天,或者每周至少5天以本文指定的时间间隔施用。该方法优选地指2型糖尿病的治疗。在公开的各种浓度范围中,优选的浓度是300U/mL。进一步,含水药物制剂还可以包括锌、间甲酚、甘油、聚山梨醇酯20和钠以及它们的混合物,它们均处于本文公开的与本发明含水药物制剂相关的范围内。在优选的实施方案中,含水药物制剂还包括0.1μg-10μg利西拉来/U甘精胰岛素。患者可以是如本文所定义的任何患者。
供制造如本文所述的以可调适的时间间隔给药的1型或2型糖尿病治疗药物的用途可以与供制造如本文所述的可减少夜间低血糖风险的1型或2型糖尿病治疗药物的用途组合。
本发明涉及,除其它事项之外,如下各项:
1.一种适用于治疗1型或2型糖尿病的含水药物制剂,其中该治疗减少夜间低血糖的风险,所述制剂包括200-1000U/mL甘精胰岛素[等摩尔于200-1000IU人胰岛素],但所述制剂的浓度不是684U/mL甘精胰岛素。
2.项1的用途限定的含水药物制剂,其包括200U/ml-650U/mL甘精胰岛素。
3.项1的用途限定的含水制剂,其包括700U/ml-1000U/mL甘精胰岛素。
4.项2的用途限定的含水制剂,其包括270-330U/mL甘精胰岛素[等摩尔于270-330IU人胰岛素]。
5.项4的用途限定的含水制剂,其包括300U/mL甘精胰岛素[等摩尔于300IU人胰岛素]。
6.前述任一项的用途限定的含水药物制剂,其中夜间低血糖选择下组:症状性低血糖、重度症状性低血糖、有记录的症状性低血糖、可能的症状性低血糖、相对症状性低血糖和无症状性低血糖。
7.前述任一项的用途限定的含水药物制剂,其中要治疗的患者在治疗开始时的HbA1c值为至少8%。
8.前述任一项的用途限定的含水药物制剂,其中要治疗的患者在治疗开始时的年龄为至少60岁。
9.前述任一项的用途限定的含水药物制剂,其中要治疗的患者在治疗开始时的BMI为至少30kg/m2。
10.前述任一项的用途限定的含水药物制剂,其中要治疗的患者在即将进行治疗之前接受过基础胰岛素。
11.前述任一项的用途限定的含水药物制剂,其中要治疗的患者在即将进行治疗之前接受过餐时短效胰岛素(餐时短效胰岛素)。
12.项10或11的用途限定的含水药物制剂,其中要治疗的患者在治疗开始时具有至少9mmol/L的注射前SMPG。
13.项10或11的用途限定的含水药物制剂,其中要治疗的患者在治疗开始时的空腹血浆葡萄糖浓度为至少8mmol/L。
14.前述任一项的用途限定的含水药物制剂,其中该制剂在夜间的预定时间每天给药一次。
15.前述任一项的用途限定的含水药物制剂,其中患者还接受餐时短效胰岛素。
16.前述任一项的用途限定的含水药物制剂,其包括毒蜥外泌肽-4类似物。
17.项16的用途限定的含水药物制剂,其中毒蜥外泌肽-4的类似物选自下组:利西拉来、艾塞那肽和利拉鲁肽。
18.项17的用途限定的含水药物制剂,其包括0.1μg-10μg利西拉来/U甘精胰岛素。
19.项18的用途限定的含水药物制剂,其包括0.2μg-1μg利西拉来/U甘精胰岛素。
20.项19的用途限定的含水药物制剂,其包括0.25μg-0.7μg利西拉来/U甘精胰岛素。
21.前述任一项的用途限定的含水制剂,其包括一种或多种选自下组的赋形剂:锌、间甲酚、甘油、聚山梨醇酯20和钠。
22.项21的用途限定的含水制剂,其包括90μg/mL锌、2.7mg/mL间甲酚和20mg/ml甘油85%。
23.项21的用途限定的含水制剂,包括90μg/mL锌、2.7mg/mL间甲酚、20μg/mL聚山梨醇酯20和20mg/ml甘油85%。
24.前述任一项的用途限定的含水药物制剂,其中pH为3.4-4.6。
25.项24的用途限定的含水制剂,其中pH是4。
26.项24的用途限定的含水制剂,其中pH是4.5。
27.项1-26中任一项的用途限定的药物制剂,其中该糖尿病是2型糖尿病。
28.项27的用途限定的药物制剂,其中该2型糖尿病仅用至少一种口服抗高血糖药未得到充分控制。
29.项28的用途限定的药物制剂,其中该至少一种口服抗高血糖药是二甲双胍。
30.项29的用途限定的药物制剂,其中用至少1.5g/天的二甲双胍治疗未充分控制该糖尿病。
31.项27-30中任一项的用途限定的含水药物制剂,其与至少一种口服抗高血糖剂组合施用。
32.项31的用途限定的含水药物制剂,其中该至少一种抗高血糖剂是二甲双胍。
33.前述任一项的用途限定的含水药物制剂,其中该制剂每天施用一次,并且其中每周有至少两天距前次施用的时间间隔在24.5h-28h的范围、或者在20h-23.5h的范围,并且其中距前次施用的平均时间间隔是大约24h。
34.一种用于治疗患者中1型或2型糖尿病的方法,包括向所述患者施用包含浓度为300U/mL的甘精胰岛素的含水药物组合物,其中该治疗减少夜间低血糖的风险。
35.项34的方法,其中所述药物组合物进一步包括选自下组的赋形剂:锌、间甲酚、甘油、聚山梨醇酯20和钠。
36.项34的方法,其中所述药物组合物进一步包括0.1μg-10μg利西拉来/U甘精胰岛素。
37.根据前述任一项的含水制剂供制造用于治疗1型糖尿病和2型糖尿病的药物的用途,其中该治疗减少夜间低血糖的风险。
38.一种适用于治疗1型或2型糖尿病的含水药物制剂,其中该制剂每天向患者施用一次,并且其中每周有至少两天距前次施用的时间间隔在24.5h-28h的范围,或者在20h-23.5h的范围,并且其中距前次施用的平均时间间隔是大约24h,所述制剂包括200-1000U/mL甘精胰岛素[等摩尔于200-1000IU人胰岛素],但所述制剂的浓度不是684U/mL甘精胰岛素。
39.项38的含水制剂,其在每周中有至少3天以项38中规定的时间间隔施用。
40.项38的含水制剂,其在每周中有至少4天以项38中规定的时间间隔施用。
41.项38-40中任一项的含水制剂,其中距前次施用的时间间隔在25h-28h的范围,或在20h-23h的范围。
42.项38-41中任一项的含水制剂,其中距前次施用的时间间隔范围在25h-27h的范围,或在21h-23h的范围。
43.项38-42中任一项的含水制剂,其中距前次施用的时间间隔范围是在25h-26.5h的范围,或在21.5h-23h的范围。
44.项38-43中任一项的用途限定的含水制剂,其包含如项2-5的任一项中限定的量的甘精胰岛素。
45.项38-44中任一项的用途限定的含水制剂,其中患者如项7-15的任一项中所限定。
46.项38-45中任一项的用途限定的含水制剂,其进一步包括如项16-20中任一项所限定的毒蜥外泌肽-4的类似物。
47.项38-46中任一项的用途限定的含水制剂,其进一步包括一种或多种如项21-23中任一项所限定的赋形剂。
48.项38-47中任一项的用途限定的含水制剂,其具有如项24-26中任一项所限定的pH。
49.项38-48中任一项的用途限定的含水制剂,其中该治疗减少夜间低血糖的风险。
50.项49的用途限定的含水制剂,其中夜间低血糖选择下组:症状性低血糖、重度症状性低血糖、有记录的症状性低血糖、可能的症状性低血糖、相对症状性低血糖和无症状性低血糖。
51.项38-50中任一项的用途限定的含水制剂,其中该糖尿病是2型糖尿病。
52.项51的用途限定的含水制剂,其中该2型糖尿病仅用至少一种口服抗高血糖剂未得到充分控制,如项28-32的任一项所限定的。
53.一种用于治疗患者中1型或2型糖尿病的方法,其中包括向所述患者施用包含浓度为300U/mL的甘精胰岛素的含水药物制剂,其中该制剂每天施用一次,并且其中每周有至少两天距前次施用的时间间隔在24.5h-28h的范围,或者在20h-23.5h的范围,并且其中距前次施用的平均时间间隔是大约24h。
54.项53的方法,其中所述药物制剂进一步包括选自下组的赋形剂:锌、间甲酚、甘油、聚山梨醇酯20和钠。
55.项53的方法,其中所述药物组合物进一步包括0.1μg-10μg利西拉来/U甘精胰岛素。
56.根据前述任一项的含水制剂供制造用于治疗1型糖尿病和2型糖尿病的药物的用途,其中该制剂每天施用一次,并且其中每周有至少两天距前次施用的时间间隔在24.5h-28h的范围,或者在20h-23.5h的范围,并且其中距前次施用的平均时间间隔是大约24h。
57.一种制品,包括包装材料、根据前述任一项的含水制剂、和表明下述内容的标签或包装材料:该制剂每天施用一次,并且其中距前次施用的时间间隔范围是21h-27h,并且其中距前次施用的平均时间间隔是大约24h。
58.一种制品,包括包装材料、根据前述任一项的含水制剂、和表明下述内容的标签或包装材料:该制剂可以和其它抗高血糖药物产品一起施用。
59.一种制品,包括包装材料、根据前述任一项的含水制剂、和表明下述内容的标签或包装材料:从每天施用一次基础胰岛素制品改为每天施用一次本制剂可以根据先前的基础胰岛素剂量单位对单位(unit-to-unit)地进行;以及从每天施用二次基础胰岛素制品改为每天施用一次本制剂,制剂的推荐初始剂量是被中断的基础胰岛素的每日总剂量的80%。
60.一种制品,包括包装材料、根据前述任一项的含水制剂、和表明下述内容的标签或包装材料:当该制剂与选自抗高血糖药品、血管紧张素转换酶(ACE)抑制剂、丙吡胺、贝特类、氟西汀、单胺氧化酶(MAO)抑制剂、己酮可可碱、丙氧芬、水杨酸盐和磺胺抗生素的可能强化该制剂的降血糖效果的物质一起施用时,制剂的剂量可能需要调整。
61.一种制品,包括包装材料、根据前述任一项的含水制剂、和表明下述内容的标签或包装材料:当该制剂与选自皮质类固醇激素、达那唑、二氮嗪、利尿药、胰高血糖素、异烟肼、雌激素和孕激素、吩噻嗪衍生物、生长激素、拟交感神经药品(例如肾上腺素[副肾素]、沙丁胺醇、特布他林),甲状腺激素、非典型抗精神病药品(例如氯氮平和奥氮平)和蛋白酶抑制剂的可能降低该制剂降血糖效果的物质一起施用时,制剂的剂量需要调整。
下文在如下附图和实施例的帮助下对本申请进行描述,它们绝非意在起限制作用。
图注
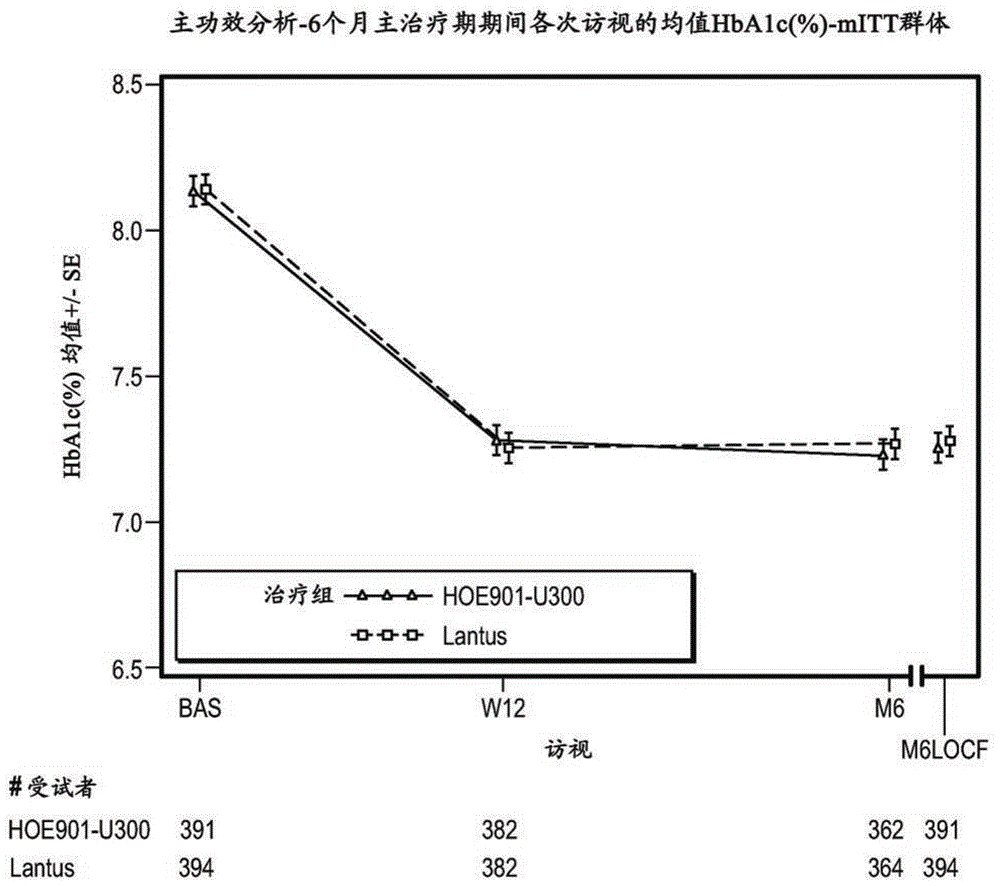
图1-主要功效分析–在6个月主治疗期期间各次访视的均值HbA1c(%)-mITT群体(实施例1)。BAS=基线,M6LOCF=6个月主治疗期期间的末次数值(LOCF)。LOCF=末次观测值结转。
图2–其它次级功效终点-在6个月主治疗期期间各次访视的均值平均注射前SMPG(mmol/L)-mITT群体(实施例1)。BAS=基线,M6LOCF=6个月主治疗期期间的末次数值(LOCF)。LOCF=末次观测值结转。
图3–其它次级功效终点–在基线和第6月终点时的均值8点SMPG概貌(mmol/l)-mITT群体(实施例1)。LOCF=末次观测值结转。
图4-其它次级功效终点-在6个月主治疗期期间各次访视的平均每日基础胰岛素和餐时胰岛素剂量(U)-mITT群体(实施例1)。BAS=基线,M6LOCF=在6个月主治疗期期间的末次数值(LOCF)。LOCF=末次观测值结转。
图5-主要功效分析-在6个月主治疗期期间各次访视的平均HbA1c(%)-mITT群体(实施例2)。BAS=基线,M6LOCF=第6月终点(LOCF),LOCF=末次观测值结转。注:对于所有在6个月期间内被救援的患者,会将救援之前和6个月治疗期间的最后一次基线后HbA1c测量值用作HbA1c终点。
图6–其它次级功效终点-在6个月主治疗期期间各次访视的均值注射前SMPG(mmol/L)-mITT群体(实施例2)。BAS=基线,M6LOCF=第6月终点(LOCF),SMPG=自我监测血浆葡萄糖,LOCF=末次观测值结转。注:对于所有在6个月期间内被救援的患者,会将救援之前和6个月治疗期间的最后一次基线后平均注射前SMPG测量值用作平均注射前SMPG终点。
图7–其它次级功效终点–在基线和第6月终点时的均值8-点SMPG概貌(mmol/l)-mITT群体(实施例2)。LOCF=末次观测值结转。SMPG=自我监测血浆葡萄糖。M6(LOCF)=第6月终点LOCF。注:对于所有在6个月期间内被救援的患者,会将救援之前和6个月治疗期间的最后一次基线后8-点概貌SMPG测量值用作8-点概貌SMPG终点。
图8–其它次级功效终点–在6个月主治疗期期间各次访视的平均每日基础胰岛素剂量(U)-mITT群体(实施例2)。BAS=基线,M6LOCF=6个月主治疗期期间末次数值(LOCF)。LOCF=末次观测值结转。注:对于所有在6个月期间内被救援的患者,会将救援之前和6个月治疗期间的最后一次基线后胰岛素剂量测量值用作胰岛素剂量终点。
图9-主要功效分析-在3个月比较方案期期间各次访视的均值HbA1c(%)-mITT子研究群体。BASM6=基线(第6月),M9LOCF=在3个月比较方案期间的末次数值(LOCF)。LOCF=末次观测值结转。
图10-在3个月比较方案期期间各次访视的平均每日基础胰岛素(甘精胰岛素)和餐时胰岛素剂量(U)-mITT子研究群体。BASM6=基线(第6月),M9LOCF=在3个月比较方案期间的末次数值(LOCF)。LOCF=末次观测值结转。
图11-在整个治疗期间平均葡萄糖(mg/dL)对一天中各小时点的作图–CGM群体。
图12-在整个晨间注射期间平均葡萄糖(mg/dL)对一天中各小时点的作图–CGM群体。
图13-在整个晚间注射期间平均葡萄糖(mg/dL)对一天中各小时点的作图–CGM群体。
图14-主要功效分析-在3个月比较方案期间各次访视的均值HbA1c(%)-mITT子研究群体。BASM6=基线(第6月),M9LOCF=在3个月比较方案期间的末次数值(LOCF)。LOCF=末次观测值结转。注:对于所有在3个月比较方案期间内被救援的患者,会将救援之前和子研究的3个月治疗期间的最后一次基线后HbA1c测量值用作HbA1c终点。
图15-在3个月比较方案期间各次访视的平均每日基础(甘精胰岛素)胰岛素剂量(U)-mITT子研究群体。BASM6=基线(第6月),M9LOCF=在3个月比较方案期间的末次数值(LOCF)。LOCF=末次观测值结转。注:所有对于在3个月比较方案期间内被救援的患者,会将救援之前和子研究的3个月治疗期间的最后一次基线后胰岛素剂量测量值用作胰岛素剂量终点。
实施例1:在2型糖尿病患者中比较甘精胰岛素新制剂与(均加用餐时胰岛素)的功效和安全性的6个月、多中心、随机、开放标签、平行分组研究,有6个月安全性延长期
概要
开发阶段:3期
目的:
主要目的:评估HOE901-U300与Lantus相比在具有餐时胰岛素的方案中作为基础胰岛素施用时的血糖控制效果,以2型糖尿病患者在6个月期间内的HbA1c变化为量度。
主要的次级目的:比较HOE901-U300和Lantus的夜间低血糖的发生率、注射前血浆葡萄糖的变化、和注射前血浆葡萄糖变异性的变化。
进一步的次级目的:
·以实现目标HbA1c值和控制血浆葡萄糖的情况为标准来比较HOE901-U300和Lantus;
·以使用糖尿病治疗满意度调查问卷(状态)(DTSQ)(在KRM中没有提供)获得的患者治疗满意度为标准来比较HOE901-U300和Lantus;
·评估HOE901-U300的安全性和耐受性(tolerability)。
方法学:以1:1(HOE901-U300对Lantus)随机化,并根据筛选时的HbA1c值进行分层(<8.0%;≥8.0%)。选择样本量(HOE901-U300 400个、Lantus 400个)以确保对主要终点(从基线到终点[6个月]的HbA1c变化)有足够的检验效能(power),并允许对第一主要的次级终点(夜间低血糖的发生率)做出结论。
患者数目:计划的:800(每个治疗臂(treatment arm)400)随机化的:807
治疗的:806
评估:疗效:804 安全性:806
诊断和纳入标准:纳入标准:根据WHO定义的2型糖尿病患者;签署书面知情同意书。关键排除标准:年龄<18岁;筛选时HbA1c<7.0%或>10%;除2型糖尿病之外的糖尿病;基础+餐时胰岛素和自我监测血糖少于1年;研究之前至少4周内每日甘精胰岛素总剂量<42U或等价的NPH剂量(如果在研究之前使用NPH作为基础胰岛素)。
研究治疗
研究的药品:测试药物:HOE901-U300;对照药物:Lantus
制剂:HOE901-U300(甘精胰岛素300U/mL溶液)是无菌、无热原、澄清、无色溶液,置于已经组装在笔式注射器的玻璃针筒中(预装填的,即一次性的笔)。Lantus(甘精胰岛素100U/mL溶液)是无菌、无热原、澄清、无色溶液,在市售的中提供(预装填的,即一次性得到笔)。
施用途径:皮下注射
剂量方案:每天夜间注射一次。注射时间在随机化时确定,并且在研究期间保持不变。
HOE901-U300或Lantus每天在夜间,即从即将晚餐前到就寝为止的任何时间,皮下注射一次。注射时间在该时间窗口内一直在相同时间,并且在随机化时由患者/研究者的裁量固定下来。患者将继续使用其餐时胰岛素类似物。
起始剂量:在基线访视之前每天使用一次Lantus或NPH的患者:HOE901-U300或Lantus的每日剂量(U)等于基线访视之前最后3天的总每日基础胰岛素剂量的中位数。
在基线访视之前使用NPH超过每日一次的患者:HOE901-U300或Lantus的每日剂量(U)比基线访视之前最后3天的总每日NPH胰岛素剂量的中位数少大约20%。
基础胰岛素剂量每周调整一次,以便使空腹SMPG到达80-100mg/dL(4.4-5.6mmol/L)的目标范围:
·如果最后3天空腹SMPG的中位数在>100mg/dL和<140mg/dL(>5.6和<7.8mmol/L)的范围内,则调整+3U
·如果最后3天空腹SMPG的中位数≥140mg/dL(≥7.8mmol/L),则调整+6U
·如果最后3天空腹SMPG的中位数在≥60mg/dL和<80mg/dL(≥3.3和<4.4mmol/L)的范围内,则调整-3U
基础胰岛素剂量优化之后,应调整餐时胰岛素剂量使血糖控制达到最佳。餐前胰岛素剂量可以随着基础胰岛素剂量的增加而减少。
非研究药品:
两个治疗组的患者在研究期间继续使用其餐时胰岛素类似物。同时使用二甲双胍治疗的患者在研究中继续使用其在研究之前接受的稳定剂量,除非由于安全性问题有必要减少剂量或中断二甲双胍。
治疗持续时间:最长达12个月
观察持续时间:最长达54周(最长达2周筛选期+6个月功效和安全性期+6个月安全性延长期+2天安全性跟踪).
功效和安全性的分析期是6个月主要治疗期。在本KRM中提供的结果是指这个时期。
评估标准:
功效:
主要功效终点:从基线到终点(第6月)的HbA1c变化。
主要的次级终点:在第9周开始与终点(第6月)之间发生至少一次夜间低血糖的患者发生率(%),标示为重度的和/或被血浆葡萄糖≤70mg/dL(3.9mmol/L)所确认;注射前SMPG从基线到终点(第6月)的变化,和注射前SMPG变异性从基线到终点(第6月)的变化。
安全性:低血糖、出现不良事件特别是治疗引发的不良事件(TEAE)和严重不良事件(SAE)、注射部位反应、和超敏反应。下列信息没有在KRM中提供:身体检查,其它安全性信息包括临床实验数据、生命体征(包括体重)、12导联ECG(12-lead ECG)和抗胰岛素抗体。
统计方法:主要功效终点(从基线到终点[6个月]的HbA1c变化)的分析使用协方差分析(ANCOVA)模型,其中以治疗、筛选HbA1c分层(<8.0和≥8.0%)和国家为固定效应,以HbA1c的基线值为协变量。HOE901-U300和Lantus之间的差异和两侧95%置信区间在ANCOVA的框架内估算。
对主要功效终点使用逐步封闭检验(stepwise closed testing)方法,以顺次评估非劣效性和优效性(superiority)。步骤1评估HOE901-U300对Lantus的非劣效性。为了评估非劣效性,将HbA1c从基线到终点的均值变化(mean change in HbA1c from baseline to endpoint)在HOE901-U300和Lantus之间的差异的双侧95%CI的上边界与HbA1c的预定非劣效性边际0.4%进行比较。如果在mITT群体上HOE901-U300和Lantus之间的差异的双侧95%CI的上边界<0.4%,则证明非劣效性。只有当证明了非劣效性时,步骤2才评估HOE901-U300对Lantus的优效性。如果在mITT群体上HOE901-U300与Lantus之间的差异的双侧95%CI的上边界<0,则证明HOE901-U300对Lantus的优效性。
只有已证明了HOE901-U300对Lantus在主要终点上的非劣性,才会在分层试验程序(hierarchical testing procedure)的框架内测试HOE901-U300对Lantus在主要的次级终点上的优效性。
概要:
群体特征:
总共807名2型糖尿病患者被随机分组到HOE901-U300(n=404)或Lantus(n=403)中;使806名患者暴露于IMP(安全性群体)。mITT群体(功效群体)包括804名患者。
总体上,每个治疗组中提前中断研究的患者的数目相当(HOE901-U300:30/404,7.4%;Lantus 31/403,7.7%)。
人口学特征和基线特征在各治疗组之间良好平衡。研究群体的平均年龄是60岁;246/807(30.4%)≥65岁。基线的均值BMI是36.6kg/m2。研究开始前的均值糖尿病持续时间是15.8年,治疗前基础胰岛素的均值持续时间是6.6年,中位数总每日胰岛素剂量是1.1U/kg体重。在两个治疗组中,基线时的均值HbA1c是8.14%。
功效结果:
主要终点:在两个治疗组中,HbA1c从基线到终点(第6月)的LS均值变化相似(HOE901-U300:-0.83%(95%CI[-0.946;-0.709]);Lantus:-0.83%(95%CI[-0.944;-0.706])。证明了HOE901-U300对Lantus的非劣效性,对Lantus的HbA1c的LS均值差异是-0.00%(95%CI[-0.112;0.107]),上边界低于预定的非劣效性边际0.4%。没有证明HOE901-U300对Lantus的优效性。
第一主要的次级终点:从第9周开始到6个月之间发生至少一次夜间重度和/或确认的低血糖的患者的发生率在HOE901-U300组[136/404(33.7%)]低于Lantus组[180/400(45.0%)]。显示了HOE901-U300对Lantus的优效性,其中相对危险度为0.75(95%CI[0.63,0.89])(p=0.0010)。
第二主要的次级终点:注射前SMPG从基线到终点(第6月)的LS均值变化在HOE901-U300(-0.90mmol/L)和Lantus组(-0.84mmol/L)中相似。治疗组之间的差异没有统计学显著性(LS平均值变化-0.06(95%CI[-0.383,0.255],p=0.6921)。
第三主要的次级终点:由于第二主要的次级终点没有证明HOE901-U300对Lantus的优效性,所以没有进一步检验第三主要的次级终点(第6月时的注射前SMPG的变异性,其在两个治疗组之间相似)。
其它次级功效终点(第6月):达到HbA1c<7%的患者比例和FPG的均值变化均在各治疗组之间相似。HOE901-U300治疗的患者与Lantus治疗的患者的8-点SMPG概貌的图形表示显示,与基线相比,终点(第6月)的血浆葡萄糖显著降低。2个治疗组的概貌在基线和终点均几乎可以重叠。
HOE901-U300组中基础胰岛素剂量的增加导致在第6月时的均值每日剂量为103U,相比之下,Lantus组的均值每日剂量为94U(两个治疗组在基线时的均值基础胰岛素剂量均为70U)。每日餐时胰岛素剂量的增加在各治疗组之间相当,在前两周有少量增加。之后,餐时胰岛素剂量保持稳定。
安全性结果:
总体上,HOE901-U300组与Lantus组相比,报告低血糖的患者的百分比始终较低。这种差异在研究治疗的前两个月以及对于夜间低血糖事件甚至更加明显。在6个月的主治疗期间,21/404(5.2%)的HOE901-U300治疗的患者报告了重度低血糖,而Lantus治疗的患者为23/402(5.7%)。
发生任何TEAE(HOE901-U300,222/404[55.0%];Lantus:215/402[53.5%])或严重TEAE(HOE901-U300,25[6.2%];Lantus,21[5.2%])的患者百分比在两组之间相似。两组中有相似比例的患者经历了严重心脏性TEAE(SOC-心脏疾病)(HOE901-U300:n=5,1.2%;Lantus:n=7;1.7%)。
研究期间有6名患者死亡,每个治疗组中3名3(0.7%)。这些人中,4名患者在前6个月期间死亡,每个治疗组2名(0.5%)。在HOE901-U300组的3名患者中,具有致命后果的事件包括如下状况:心脏的感染性血栓和栓塞,支气管肺癌具转移,以及—对于第三名患者—肺栓塞。Lantus组的3名患者中致命事件的主要原因包括:长期抑郁和药物中毒,1名患者具有多种状况,包括慢性心脏衰竭(NYHA IV)的恶化,慢性肾功能衰竭4期急性失代偿,促成致命后果的失代偿性糖尿病和糖尿病肾病,最后1名患者罹患病因不明的急性心跳呼吸骤停。没有死亡被认为与研究药物有关。
两个治疗组中有相似数目的患者经历了导致永久性治疗中断的TEAE(HOE901-U300:n=6,1.5%;Lantus:n=7,1.7%)。
在6个月主治疗期期间报告的超敏反应在两个治疗组中相似(HOE901-U300:n=3,0.7%;Lantus:n=2,0.5%)。
总体上,6个月主治疗期期间的注射部位反应在两个治疗组中的比例相似(HOE901-U300:n=9,2.2%;Lantus:n=6,1.5%)。
结论:
在本研究的807名依靠基础胰岛素联合餐时胰岛素的T2DM患者中,各治疗组之间的基线特征和人口特征得到良好平衡。对于主要功效终点(从基线到终点[6个月]的HbA1c变化),显示了HOE901-U300对Lantus的非劣效性。对于从第9周开始到第6月之间报告了夜间低血糖(以SMPG≤70mg/dL[3.9mmol/L]作为重度和/或被确认的指示)的患者的发生率(%),HOE901-U300组显著低于Lantus组(分别为33.7%和45%,RR为0.75,p-值为0.0010;第一主要次级功效终点)。对于其它次级终点,例如注射前血浆葡萄糖、注射前血浆葡萄糖的变异性、达到目标HbA1c的患者数目、FPG的平均值变化和血浆葡萄糖的8-点概貌,各治疗组之间的结果相当。
在6个月主治疗期期间低血糖的总发生率(发生至少一次事件的患者%),HOE901-U300组低于Lantus组,而与低血糖的范畴无关。
HOE901-U300在研究的6个月主治疗期期间得到了良好耐受,没有观察到特定的安全性问题。
12个月EDITION 1延长研究的功效和安全性结果概要
-HbA1c:在安全性延长期(从主要研究终点[6个月]到治疗结束[第12月])期间,HbA1c保持稳定并且在两个治疗组中相当。
-低血糖:总体上,与6个月主治疗期期间相似,在整个研究治疗期期间,HOE901-U300组与Lantus组相比,发生低血糖的患者百分数相似或更低。
-安全性:HOE901-U300在研究期间良好耐受,并且没有观察到特定的安全性问题;在整个治疗期间,两个组中发生任何TEAE的患者的百分比相似(HOE901-U300为289/404[71.5%],Lantus治疗组为278/402[69.2%]),且没有特定的SOC贡献。报道严重TEAE的患者的数目相似:(HOE901-U300为53[13.1%]),Lantus治疗组为62[15.4%])。HOE901-U300中有2位(0.5%)患者和Lantus治疗组中有4位(1.0%)患者在整个研究治疗期间发生TEAE并导致死亡。
-体重:在两个治疗组中,在整个研究治疗期间,体重有少量的增加(HOE901-U300是1.17kg,Lantus是1.40kg)。
1.结果
1.1研究患者
1.1.1研究处理(Study disposition)
表1–患者处置-随机化群体
注意:百分比用经随机化的患者的数目作为分母计算
表2–分析群体
注意:对于安全性群体,患者根据实际接受的治疗(依治疗)作表
对于其它群体,患者根据其随机化分配的治疗作表。
1.1.2人口特征(Demographics)和基线特征
表3-人口特征和患者基线特征–随机化群体
BMI=体重指数
GFR=肾小球滤过率
GFR是从MDRD公式导出的。
表4–疾病基线特征的汇总–随机化群体
T2D=2型糖尿病
a患者的最大注射数目
b在随机化之前至少7天期间患者基础/餐时/总每日剂量的平均值
c在筛选之前3个月内获得
1.2功效评估
1.2.1主要功效终点
表5–主要功效分析–使用LOCF程序从基线到第6月终点的HbA1c(%)均值变化-mITT群体(图1)
LOCF=末次观测值结转
a处理组(HOE901-U300和LANTUS)的协方差分析(ANCOVA)模型,筛选HbA1c的随机化分层(<8.0,≥8.0%),国家作为固定效应,基线HbA1c值作为协变量
1.2.2主要次级终点
1.2.2.1夜间低血糖
表6-第一主要次级功效终点–在第9周开始至第6月终点之间(使用LOCF程序)发生至少一次夜间低血糖[00:00-05:59]的患者数目(%),以血浆葡萄糖≤3.9mmol/L(70mg/dL)作为重度和/或被确认的指示-mITT群体
n(%)=发生至少一次夜间低血糖事件的患者数目和百分比,指示为重度和/或为血浆葡萄糖≤3.9mmol/L(70mg/dL)所确认
a根据筛选HbA1c的随机化分层(<8.0或≥8.0%)分层的RR,使用CMH方法
1.2.2.2注射前血浆葡萄糖–第6月终点
表7–第二主要次级功效终点-使用LOCF程序从基线到第6月终点的平均注射前SMPG(mmol/L)的均值变化-mITT群体(图2)
LOCF=末次观测值结转
SMPG=自我监测血浆葡萄糖
a平均值用给定访视之前7天内计算的至少3次SMPG的平均值进行估算
b方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层(<8.0,≥8.0%)、和国家作为固定效应,以基线平均注射前SMPG值作为协变量
在V1(第2周),研究者或研究助理将给患者提供血糖仪和相应配件(针头、对照溶液、试纸条等)以及日记本,以执行血浆葡萄糖自我测量和记录。将给患者展示如何用血糖仪准确测量血浆葡萄糖。研究者或研究助理将说明在概貌所需的时刻测量葡萄糖、并正确地将数值记录在患者日记中的必要性。根据需要的频度在各次研究访视时重复训练,并且由研究助理在每次访视时检查患者日记。由患者使用赞助者提供的血糖仪测量血糖值。患者将把他们的SMPG数据记录在日记中。
嘱咐患者每次前往接受访视时携带赞助者提供的血糖仪。血糖仪应该按照包装册页中提供的使用说明校准,并且研究点还应当使用所提供的对照溶液定期检查血糖仪,以保证数据的有效性。
从V1(筛选访视)开始,日记包括由患者记录的部分:
·研究治疗和餐时胰岛素:HOE901-U300或Lantus注射的时间和剂量(筛选期期间的Lantus或NPH注射)和餐时/餐前胰岛素类似物注射;
·SMPG:SMPG的时间和数值;
1.晨间(早餐前)的空腹SMPG
2.在每次访视前7天内在注射基础胰岛素之前30分钟的SMPG
3.SMPG的4-点概貌和8-点概貌
4.与低血糖事件相关的SMPG
5.任何其它SMPG(无论测量的原因如何)
SMPG测量的时刻安排如下:
·空腹血浆葡萄糖(SMPG):空腹(早餐前)SMPG在整个研究期间每天测量。在第3访视(第1天,基线)之前的一周内,根据是否遵守空腹SMPG测量时刻表来评估是否适格纳入随机治疗期。在研究期间,当完成增大剂量(uptitration)并且空腹(早餐前)SMPG稳定在目标范围内时,每周应当至少执行3次测量。
·注射研究药物前30分钟内的血浆葡萄糖(注射前SMPG):,在基线之前的至少7天中,以及在整个研究中的每次现场访视(site visit)之前,在注射IMP(HOE901-U300或Lantus)之前30分钟内完成注射前SMPG。在完成4-点或8点概貌的各天里:如果注射前SMPG的时间与4-点或8点概貌的某个时间点相同,则患者在日记中必须对两者都赋予SMPG值(例如,注射前PG;就寝时)。
·4点SMPG概貌(早餐前,午餐前,晚餐前,就寝前):在基线访视之前的1周内和在用IMP治疗的前12周内,患者每周至少有3天执行4点SMPG概貌。一旦达到滴定目标,可以根据研究者的判断减少4点SMPG概貌的次数,但是每次访视前1周内必须至少有3天执行4点SMPG概貌。然而,推荐在整个研究期间每天执行4点SMPG概貌,以便对胰岛素方案作出最佳判断;
·8点血糖概貌(从夜间03:00am测量开始;早餐前和早餐后2小时;午餐前和午餐后2小时;晚餐前和晚餐后2小时;就寝时):患者在每次现场访视之前的5天内至少有一天执行8-点SMPG概貌。不过,推荐每周至少执行一次8-点SMPG概貌直至第8周,之后每两周执行一次。特别需要注意的是记录3.00a.m.SMPG值。
·症状性低血糖发作期间的SMPG:无论何时患者感到有低血糖症状,如果可能的话,患者(或者他人,如果可以)均应当测量血浆葡萄糖。无论何时怀疑有症状性低血糖,应嘱咐患者在给予葡萄糖或糖类之前测量血浆葡萄糖水平,除非由于安全性考虑必须在确认之前立即进行葡萄糖/糖类救援。
下面的SMPG值必须拷贝到eCRF中:
在基线访视前1周和前12周内,直至第8次访视(12周):
·空腹(早餐前)SMPG:每天
·注射前SMPG(在注射基础胰岛素之前30分钟内):在每次现场访视前的7天
·4-点概貌SMPG:每周的不同的3天,直至第8次访视(12周)
·8-点概貌SMPG:每次现场访视之前的5天内执行1次概貌
·与低血糖事件有关的SMPG:任何被记录的时刻
注意:在电话访视之后,至少将下面的数据将输入到e-CRF中:最近3天的空腹(早餐前)SMPG,与低血糖事件有关的SMPG:任何被记录的时刻。电话访视前1周的其余SMPG数据将在随后的现场访视中输入到e-CRF中。
第8次访视之后(第12周):
·空腹(早餐前)SMPG:每次现场访视之前的7天内
·注射前SMPG(注射基础胰岛素之前30分钟内):每次现场访视之前7天时
·4-点概貌SMPG:在每次现场访视之前的7天内的不同3天
·8-点概貌SMPG:在每次现场访视之前的5天内的一天
·与低血糖事件有关的SMPG:任何被记录的时刻
所有葡萄糖数值将被研究者用于监视血糖。
1.2.2.3注射前SMPG的变异性-第6月终点
表8–第三主要的次级功效终点-使用LOCF程序从基线至第6月终点的注射前SMPG变异性的均值变化-mITT群体
LOCF=末次观测值结转.
SMPG=自我检测血浆葡萄糖
变异性用给定访视之前的7天内测量的至少3次SMPG计算的变异系数的均值进行估算
a协方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层(<8.0,≥8.0%)、以及国家作为固定效应
1.2.3其它次级的功效终点
1.2.3.1在6个月时HbA1c<7%的患者百分比
表9–其它次级功效终点-第6月终点时HbA1c<7%的患者的数目(%)(使用LOCF程序)和第6月终点时HbA1c<7%并且在6个月主治疗期的最后3个月期间没有经历以被指示为重度和/或被血浆葡萄糖<3mmol/L(54mg/dL)所确认的低血糖的患者的数目(%)(使用LOCF程序)–mITT群体
LOCF=末次观测值结转.
RR=相对危险度
a根据筛选HbA1c的随机化分层(<8.0或≥8.0%)分层的RR,使用CMH方法
1.2.3.2 FPG从基线至第6月的变化
表10–其它次级功效终点–使用LOCF程序从基线至第6月终点FPG(mmol/L)的均值变化-mITT群体
FPG=空腹血浆葡萄糖
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层(<8.0,≥8.0%)和国家作为固定效应,基线FPG值作为协变量
1.2.3.3八-点SMPG概貌
图3描述了基线和第6月终点时的平均八-点SMPG概貌(mmol/l)——mITT群体。
1.2.3.4.基础和餐时胰岛素剂量
图4描述了6个月主治疗期期间通过访视获得的平均每日基础胰岛素和餐时胰岛素剂量(U)-mITT群体。
1.3安全性评估
1.3.1暴露的程度
表11–在6个月主治疗期期间暴露于研究产品-安全性群体
注:患者视为处于他们在随机化时被实际分配到的治疗组中
1.3.2低血糖
表12在6个月主治疗期期间发生至少一次突发低血糖事件的患者数目(%)-安全性群体
n(%)=发生至少一次低血糖事件的患者数目和百分比
a确认的低血糖=有记录的症状性低血糖或无症状低血糖
低血糖事件按照如下进行分类(American Diabetes Association Workgroup on Hypoglycemia.Defining and Reporting Hypoglycemia in Diabetes.Diabetes Care 2005;28:1245-49):
重度低血糖
重度低血糖是需要他人帮助才能主动给予糖类、胰高血糖素或其它救援行动的事件。
这些事件可能伴随足以诱发抽搐、意识障碍或昏迷的神经低血糖症。在此类事件期间可能无法进行血浆葡萄糖测量,但是由于血浆葡萄糖恢复正常所导致的神经恢复被认为足以证明该事件是由于低血浆葡萄糖浓度所诱导的。
重度症状性低血糖的定义可包括所有神经障碍严重到足以阻止自我治疗,并因此被认为将患者置于伤害自身或他人的风险之中的情况。
注意,“需要帮助”意思是指患者不能自助。在不需要帮助时,出于善意地帮助患者不应该被认为是“需要帮助”事件。
重度症状性低血糖只有当满足SAE标准时才构成SAE。所有抽搐、意识障碍或昏迷的事件都必须作为SAE报告。
有记录的症状性低血糖
有记录的症状性低血糖是这样的事件:事件期间典型的低血糖症状伴随测得血浆葡萄糖浓度≤70mg/dL(≤3.9mmol/L)(American Diabetes Association Workgroup on Hypoglycemia.Defining and Reporting Hypoglycemia in Diabetes.Diabetes Care 2005;28:1245-49)。
被认为由于低血糖发作导致的临床症状包括,例如:多汗、精神紧张、乏力/虚弱、震颤、头晕、食欲增加、心悸、头痛、睡眠障碍、精神错乱、抽搐、神志不清、昏迷。
无症状低血糖
无症状低血糖是不伴随典型低血糖症状,但是测得的血浆葡萄糖浓度小于或等于70mg/dL(3.9mmol/L)的事件。
可能的症状性低血糖
可能的症状性低血糖是这样的事件,事件期间低血糖症状并不伴随血浆葡萄糖确定,但是被假定是由于血浆葡萄糖浓度小于或等于70mg/dL(3.9mmol/L),症状用口服糖类治疗,而没有测量血浆葡萄糖。
相对低血糖
相对低血糖是这样的事件,事件期间糖尿病患者报告有任何典型的低血糖症状,并且将该症状解释为低血糖的指示,但是测得的血浆葡萄糖浓度大于70mg/dL(3.9mmol/L)。
夜间低血糖
夜间低血糖是在00:00-05:59a.m.之间发生的上述类型的任何低血糖。注意:在主要的次级终点分析(发生至少一次夜间低血糖的患者)中不会包括相对夜间低血糖。
除了小于或等于70mg/dL(3.9mmol/L)的阈值之外,血浆葡萄糖<54mg/dL(3.0mmol/L)的低血糖发作将被单独分析(Guideline on clinical investigation of medicinal products in the treatment of diabetes mellitus.Draft.EMA,2010年1月20日)。
低血糖的分类根据钟表时间进行:
低血糖事件按其日分布(diurnal distribution)(0:00-24:00)此外按每日中时间进行分析:
ο按每日中时间定义的夜间低血糖:在00:00-05:59a.m.之间发生的任何上述类型的低血糖,不管患者是清醒的还是由于该事件而醒来;
ο日间低血糖:在6:00a.m.-23:59之间发生的任何上述类型的低血糖。
都将嘱咐患者无论任何时候怀疑有症状性低血糖时,在给予糖类之前都应测量手指穿刺血浆葡萄糖水平,除非由于安全性考虑必须在确认之前立即进行葡萄糖救援,并且然后在安全前提下应当尽快进行葡萄糖测量,并合适地进行日志记录。
关于低血糖事件的细节将被捕捉在患者日记中,并且患者在严重事件之后将尽快联系研究站,以审阅细节并确定要采取的任何必要的措施。
所有低血糖发作均将被记录在e-CRF的“低血糖具体表格(hypoglycemia specific form)”中。这包括所有症状性低血糖事件和无症状低血糖。满足SAE标准的低血糖事件将被记录在e-CRF的SAE表格中。
对每位患者,每个患者年的低血糖发生率可以计算为:365.25x(低血糖发作的次数)/(暴露的天数),并且按照事件类型和治疗组进行总结。
表13在6个月主治疗期内的各研究期发生至少一次突发低血糖事件的患者数目(%)-安全性群体
n(%)=发生至少一次低血糖事件的患者数目百分比
a确认的低血糖=有记录的症状性低血糖或无症状低血糖
1.3.3治疗引发的不良事件
表14–在6个月治疗期间的治疗引发的不良事件-安全性群体
TEAE:治疗引发的不良事件,SAE:严重不良事件
n(%)=发生至少一次TEAE的患者的数目和百分比
表15–在6个月主治疗期期间在任何治疗组中按主要SOC、HLT和PT区分的发生了HLT≥2%的TEAE的患者数目(%)-安全性群体
TEAE:治疗引发的不良事件,SOC:系统器官分类(System organ class),HLT:高级别术语(High level term),PT:优选术语(Preferred term)MedDRA 15.1
n(%)=发生至少一次TEAE的患者的数目和百分比
注:表格排序是按照SOC国际公认的顺序,HLT、PT按照字母顺序
只提供了在至少一个组中有至少一个<HLT≥2%>的HLT
1.3.4死亡、重度治疗引发的不良事件
1.3.4.1死亡
表16–在研究各期间(研究期,治疗期,研究后)死亡的患者数目(%)-安全性群体
TEAE:治疗引发的不良事件,SAE:严重不良事件
a包括从治疗开始到研究结束(定义为最后一次根据方案计划的访视或所有预定监视的治疗引发SAE和不良事件的解决/稳定化)时发生的所有死亡
b治疗期是6个月主治疗期
c包括在研究结束之后(如脚注a所定义)发生、并在数据库中报告的死亡
1.3.4.2严重不良事件
表17–在6个月主治疗期期间按照主要SOC、HLGT、HLT和PT区分的发生了治疗引发SAE的患者数目(%)-安全性群体
SAE:严重不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语MedDRA 15.1
n(%)=发生至少一次治疗引发SAE的患者数目和百分比
注:表格根据SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
1.3.5导致退出的不良事件
表18–根据主要SOC,HLGT,HLT和PT区分的在6个月治疗期间发生导致永久性治疗中断的TEAE的患者的数目(%)-安全性群体
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语
MedDRA 15.1
n(%)=发生至少一次导致永久性治疗中断的TEAE的患者数目和百分比
注:表格根据SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
1.3.6.其它显著的不良事件
1.3.6.1超敏反应
表19–根据相关标准化MedDRA问卷和优选术语–超敏反应区分的在6个月主治疗期期间发生了至少一次TEAE的患者数目(%)-安全性群体
MedDRA 15.1
TEAE:治疗引发的不良事件
n(%)=发生至少一次超敏反应事件的患者数目和百分比
1.3.6.2注射部位反应
表20–根据相关标准化MedDRA问卷和优选术语–注射部位反应区分的在6个月主治疗期期间发生了至少一次TEAE的患者数目(%)-安全性群体
MedDRA 15.1
TEAE:治疗引发的不良事件
n(%)=在注射部位事件中发生至少一次局部耐受性的患者数目和百分比
实施例2:在2型糖尿病患者中比较甘精胰岛素新制剂与的功效和安全性的6个月、多中心、随机、开放标签、平行分组研究,两组均联用口服抗高血糖药物,并有6个月安全性延长期
概要
研究中心:多中心
开发阶段:3
目的:
主要目的:评估HOE901-U300在与口服抗高血糖药物联合的方案中作为基础胰岛素施用时与Lantus相比对血糖控制的效果,以2型糖尿病患者在6个月期间内的HbA1c变化为量度。
主要的次级目的:比较HOE901-U300和Lantus的下列方面:夜间低血糖的发生率、注射前血浆葡萄糖的变化、和注射前血浆葡萄糖变异性的变化。
进一步的次级目的:
·以达到目标HbA1c值和可控的血浆葡萄糖为量度来比较HOE901-U300和Lantus;
·以使用糖尿病治疗满意度调查问卷(状态)(DTSQ)(在KRM中没有提供)获知的患者治疗满意度为量度来比较HOE901-U300和Lantus;
·评估HOE901-U300的安全性和耐受性(tolerability)。
方法学:以1:1(HOE901-U300与Lantus)随机化,并根据筛选时的HbA1c值进行分层(<8.0%;≥8.0%)。选择样本量(400人使用HOE901-U300,400人使用Lantus)以确保对主要终点(HbA1c从基线到终点[第6月]的变化)有足够的效能,并允许对第一主要次级终点(夜间低血糖的发生率)做出结论。
患者数目:计划:800(每个治疗臂(treatment arm)400)随机化:811
治疗:809
评估:功效:808 安全性:809
诊断和纳入标准:纳入标准:在筛选访视时已根据WHO定义被诊断为2型糖尿病至少1年的患者;签署书面知情同意书。关键排除标准:年龄<18岁;筛选时HbA1c<7.0%或>10%;除2型糖尿病之外的糖尿病;基础胰岛素治疗联合口服抗高血糖剂及血糖自我监测少于6个月;研究之前最后4周内总每日甘精胰岛素剂量<42U或NPH的等价剂量(如果在研究之前使用NPH作为基础胰岛素)。
研究治疗
研究的药品:测试药物:HOE901-U300;对照药物:Lantus
制剂:HOE901-U300(甘精胰岛素300U/mL溶液)是无菌、无热原、澄清、无色溶液,置于已经组装在笔式注射器的玻璃针筒(预装填的,即一次性的笔)中。Lantus(甘精胰岛素100U/mL溶液)是无菌、无热原、澄清、无色溶液,作为(预装填的,即一次性的笔)在市场上销售。
施用途径:皮下注射
剂量方案:每天夜间注射一次。注射时间在随机化时固定,并且在研究期间保持不变。
起始剂量:在基线访视之前已经每天使用一次Lantus或NPH的患者:HOE901-U300或Lantus的每日剂量(U)等于基线访视之前最后3天总每日基础胰岛素剂量的中位数。
在基线访视之前超过每日一次NPH的患者:HOE901-U300或Lantus的每日剂量(U)比基线访视之前最后3天总每日NPH胰岛素剂量的中位数少大约20%。
基础胰岛素剂量每周调整一次,以使空腹SMPG达到80-100mg/dL(4.4-5.6mmol/L)的目标范围:
·如果最后3天空腹SMPG的中位数在>100mg/dL和<140mg/dL(>5.6和<7.8mmol/L)的范围内,则调整+3U
·如果最后3天空腹SMPG的中位数≥140mg/dL(≥7.8mmol/L),则调整+6U
·如果最后3天空腹SMPG的中位数在≥60mg/dL和<80mg/dL(≥3.3和<4.4mmol/L)的范围内,则调整-3U
救援治疗:
如果在12周或者之后,基础胰岛素调整不能将FPG/HbA1c降低到11.1mmol/L(200mg/dL)的FPG阈值和8%的HbA1c阈值之下,并且没有确认明显的控制不充分的理由,则需要考虑增加治疗强度。根据研究者的判断和本地标签文件选择在基础胰岛素和口服抗高血糖背景疗法的基础上增加的抗糖尿病治疗。
非研究药品:
两个治疗组的患者在研究期间继续使用其稳定剂量的口服抗高血糖背景疗法,但是磺酰脲在筛选访视之前2个月内和研究期间是禁用的。救援疗法也被视为非研究药品。
治疗持续时间:最长达12个月
观察持续时间:最长达58周(长达2周筛选期+6个月功效和安全性期+6个月安全性延长期+4天治疗后跟踪期).
功效和安全性的分析期是6个月主治疗期。在本KRM中提供的结果是指这个时期。
对于所有在6个月治疗期内需要救援治疗的患者,将开始救援治疗之前的最后一次基线后功效测量值用作功效终点。这些患者在开始救援治疗之后被排除在功效分析之外。对于安全性终点;分析期是6个月主治疗期,而不管是否使用救援疗法。
评估标准:
功效:
主要功效终点:HbA1c从基线到终点(第6月)的变化。
主要的次级终点:从第9周开始到终点(第6月)之间发生至少一次被指示为重度和/或被血浆葡萄糖≤70mg/dL(3.9mmol/L)所确认的夜间低血糖的患者的发生率(%);从基线到终点(第6月)之间的注射前SMPG变化,和从基线到终点(第6月)之间的注射前SMPG变异性的变化。
安全性:低血糖,出现不良事件特别是治疗引发的不良事件(TEAE)和严重不良事件(SAE),导致退出的TEAE和导致死亡的TEAE,注射部位反应和超敏反应。下列信息没有在KRM中提供:身体检查,其它安全性信息包括临床实验数据、生命体征(vital sign)(包括体重)、12导联ECG(12-lead ECG)和抗胰岛素抗体。
下面的信息没有在本KRM中提供:身体检查,其它安全性信息包括临床试验数据、生命体征、12导联ECG和抗胰岛素抗体。
统计方法:主要功效终点(HbA1c从基线到终点[第6月]的变化)的分析使用协方差分析(ANCOVA)模型,以治疗、筛选HbA1c分层(<8.0和≥8.0%)和国家为固定效应,以HbA1c的基线值为协变量。HOE901-U300和Lantus之间的差异和两侧95%置信区间在ANCOVA的框架内估算。
主要功效终点使用逐步封闭检验方法(stepwise closed testing approach)以顺次评估非劣效性和优效性。步骤1评估HOE901-U300对Lantus的非劣效性。为了评估非劣效性,将HOE901-U300和Lantus之间从基线到终点的HbA1c平均值变化差异的双侧95%CI的上边界与HbA1c的预定非劣效性边际0.4%进行比较。如果HOE901-U300和Lantus之间在mITT群体上的差异的双侧95%CI的上边界<0.4%,则证明非劣效性。只有当非劣性得到证明时,步骤2才评估HOE901-U300对Lantus的优效性。如果HOE901-U300和Lantus之间在mITT群体上的差异的双侧95%CI的上边界<0,则证明HOE901-U300对Lantus的优效性。
只有对主要终点证明了HOE901-U300对Lantus的非劣效性,才会在分层检验程序(hierarchical testing procedure)的框架内对主要的次级终点进行HOE901-U300对Lantus的优效性检验。安全性分析的描述系基于安全性群体。
概览:
群体特征:
总共811名2型糖尿病患者被随机分组到HOE901-U300(n=404)或Lantus(n=407)中;809名患者暴露于IMP(安全性群体)。mITT群体(功效群体)包括808名患者。
总体上,每个治疗组有相当数目的患者提前中断了研究治疗(HOE901-U300:36/404,8.9%;Lantus 38/407,9.3%)。HOE901-U300臂中总共有344名(85.1%)患者、Lantus臂中有349名(85.7%)患者完成了6个月主治疗期(接受救援治疗的患者被排除在完成者群体之外)。
人口学特征和基线特征在各治疗组之间良好平衡。研究群体的均值年龄是58.2岁;190/811(23.4%)≥65岁。基线的均值BMI是34.8kg/m2。HOE901-U300组BMI超过40kg/m2的患者(21.5%)与Lantus组(16.7%)相比略多。研究开始前的平均糖尿病持续时间是12.6年,治疗前基础胰岛素的平均持续时间是3.8年。在研究治疗开始前7天,大部分患者使用甘精胰岛素(78.8%相对NPH 21.2%);Lantus组中使用甘精胰岛素的患者(82.8%)比HOE901-U300组(74.9%)多。基线时中位数总每日胰岛素剂量是0.614U/kg体重。
两个治疗组的基线均值HbA1c相似(HOE901-U300:8.28%,Lantus:8.22%;针对可评估的患者,即具有基线和至少一个基线后HbA1c评估的患者)。
功效结果:
主要终点:在两个治疗组中,HbA1c从基线到终点(第6月)的LS平均值变化相似(HOE901-U300:-0.57%(95%CI[-0.756;-0.387]);Lantus:-0.56%(95%CI[-0.744;-0.379])。证明了HOE901-U300对Lantus的非劣效性,Lantus的HbA1c的LS均值差异是-0.01%(95%CI[-0.139;0.119]),上边界低于预定的非劣效性边际0.4%。在使用0.3%的非劣效性边际时,非劣效性亦成立。没有证明HOE901-U300对Lantus的优效性。
第一主要的次级终点:从第9周开始到第6月之间发生至少一次夜间重度和/或确认的低血糖的患者的发生率,HOE901-U300组[87/403(21.6%)]低于Lantus组[113/405(27.9%)]。显示了HOE901-U300对Lantus的优效性,相对危险度为0.77(95%CI[0.61,0.99])(p=0.0380)。
第二主要的次级终点:注射前SMPG从基线到终点(第6月)的LS平均值变化,在HOE901-U300(-0.56mmol/L)和Lantus组(-0.51mmol/L)中相似。治疗组之间的差异没有统计学显著性(LS平均值变化-0.04(95%CI[-0.438,0.350],p=0.8279)。
第三主要的次级终点:因为第二主要的次级终点没有证明HOE901-U300对Lantus的优效性,所以没有对第三主要的次级终点进行进一步的检验(在第6月时注射前SMPG的变异性降低,HOE901-U300组(-2.34)在数值上比Lantus组(-0.53)更大)。
其它次级功效终点(第6月):治疗组之间达到HbA1c<7%的患者比例和FPG的平均值变化均相似(HOE901-U300是30.6%;Lantus是30.4%)。两个治疗组显示FPG平均值变化相似的降低。两个治疗组的8-点SMPG概貌的图形表示显示,与基线相比,终点(第6月)的血浆葡萄糖显著降低。
在第6月,HOE901-U300组的均值每日胰岛素剂量是91U(0.92U/kg),Lantus组是82U(0.84U/kg)。
在6个月主治疗期期间,两个治疗组中有相似数目的患者接受了救援治疗(HOE901-U300是5.7%;Lantus是4.9%)。
安全性结果:
总体上,HOE901-U300组与Lantus组相比,报告低血糖的患者百分比一贯较低。这种差异在研究治疗的前两个月中以及对于夜间低血糖事件而言尤为明显。在6个月主治疗期期间,4/403(1%)的HOE901-U300治疗患者、以及6/406(1.5%)的Lantus治疗患者报告了重度低血糖。
发生任何TEAE(HOE901-U300,236/403[58.6%];Lantus:206/406[50.7%])的患者百分比HOE901-U300组高于Lantus组,没有特定的SOC贡献。两个治疗组有相似数目的患者报告了严重TEAE(HOE901-U300,15[3.7%];Lantus,15[3.7%])。
6个月治疗期间HOE901-U300组有2名(0.5%)患者死亡,Lantus治疗组有1名(0.2%)患者死亡。
在HOE901-U300组的2名患者中,具有致命后果的事件包括由于晚期冠状动脉病症导致的心肌梗死和心源性猝死。两位患者均患有预先存在的显著心血管病理,并具有多种导致致命后果的风险因素。Lantus组的患者经历了导致死亡后果的慢性肾盂肾炎急性发作。在6个月治疗期间发生的死亡无一被认为与研究药物有关。
两个治疗组中有相似数目的患者经历了导致永久治疗中断的TEAE(HOE901-U300:n=6,1.5%;Lantus:n=4,1.0%)。
在6个月主治疗期期间报告的超敏反应的比例在两个治疗组中相似(HOE901-U300:n=13,3.2%;Lantus:n=16,3.9%)。
总体上,6个月主治疗期期间报告的注射部位反应的比例,Lantus治疗组高于HOE901-U300组(Lantus:n=12,3.0%;HOE901-U300:n=4,1.0%)。
在两个组中体重均无明显变化(HOE901-U300为0.08kg,Lantus为0.66kg)。
结论:
在本研究的811名依靠基础胰岛素联合口服抗高血糖药物的T2DM患者中,治疗组之间的基线特征和人口学特征良好平衡。对于主要功效终点(从基线到终点[第6月]的HbA1c变化),HOE901-U300对Lantus显示了非劣效性。从第9周开始到第6月之间报告了夜间低血糖(以SMPG≤70mg/dL[3.9mmol/L]作为重度和/或被确认的指示)的患者发生率(%),HOE901-U300组显著低于Lantus组(分别为21.6%和27.9%,RR为0.77,p-值为0.0380;第一主要次级功效终点)。对于其它次级终点,如注射前血浆葡萄糖、注射前血浆葡萄糖的变异性、达到目标HbA1c的患者数目、FPG的平均值变化和8-点SMPG的变化,和24小时平均血浆葡萄糖的变异性,发现治疗组之间的结果相当。
在6个月主治疗期期间低血糖的总发生率(发生至少一次事件的患者%),HOE901-U300组一贯低于Lantus组,而与低血糖的类型无关。这种有利于HOE901-U300的差异对于所有分类的夜间低血糖尤为显著。
HOE901-U300在6个月研究主治疗期间被良好耐受,没有观察到特定的安全性问题。
12个月的EDITION 2延长研究的功效和安全性结果汇总
-HbA1c:在安全性延长期(从主要研究终点[第6月]到治疗结束[第12月])中,HbA1c保持稳定并且在两个治疗组中相当。
-救援:在整个治疗期间,需要救援治疗的患者百分比在两个治疗组中相似(HOE901-U300是8.2%;Lantus是10.1%)。在6个月安全性延长期内开始救援治疗的患者百分数HOE901-U300治疗组低于Lantus组(HOE901-U300是2.5%;Lantus是5.4%)。
-低血糖:总体上,与6个月主治疗期期间相似,在整个研究治疗期间,HOE901-U300组与Lantus组相比,发生低血糖的患者百分数更低,且与低血糖分类无关。
-安全性:HOE901-U300在研究期间良好耐受,并且没有观察到特定的安全性问题;HOE901-U300治疗组中在整个治疗期间发生了任何TEAE的患者百分数高于Lantus组(分别是278/403[69.0%]和244/406[60.1%]),没有特定的SOC贡献。两个治疗组中报道严重TEAE的患者数目相似。HOE901-U300中有4位(1%)患者和Lantus治疗组中有2位(0.5%)患者在整个研究治疗期间发生了导致死亡的TEAE。
-体重:在两个治疗组中,在整个研究治疗期间体重有少量增加(对于HOE901-U300是0.41kg,对于Lantus是1.15kg)。
2.结果
2.1研究患者
2.1.1研究处置
表21–患者处置–随机化群体
注:百分比使用随机患者的数目作为分母计算
完成6个月主治疗期的患者是没有永久性中断研究治疗并且没有使用任何救援药物的患者
aLantus臂中的2个受试者在截止日期之后获得其研究状态
表22–分析群体
注:对于安全性群体,患者根据其实际接受的治疗(根据治疗)列表
对于其它群体,患者根据其被随机化的治疗列表
表23–在6个月主治疗期期间需要救援疗法的患者数目(%)-mITT群体
RR=相对危险度
a根据筛选HbA1c的随机化分层(<8.0或≥8.0%)分层的RR,使用CHM方法
2.1.2人口学特征和基线特征
表24–基线处的人口学特征和患者表征-随机化群体
BMI=体重指数
GFR=肾小球滤过率
GFR是从MDRD公式导出的
表25–基线处的病症特征概要-随机化群体
T2D=2型糖尿病
a在随机化之前的最后7天内,患者的先前基础胰岛素类型和最大注射数目
b在随机化之前的最后7天内,患者基础每日剂量的平均值
c在筛选之前的3个月获取
2.2功效评估
2.2.1主要功效终点
表26-主要功效分析-使用LOCF程序从基线至第6月终点的HbA1c(%)平均值变化-mITT群体
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层(<8.0,≥8.0%)和国家作为固定效应,以基线HbA1c值作为协变量
注:对于6个月期间的所有被救援患者,救援前和6个月治疗期间的最后一次基线后HbA1c测量结果用作HbA1c终点
表26的数据总结在图5中。
2.2.2主要的次级终点
2.2.2.1夜间低血糖
表27–第一主要的次级功效终点–在从第9周开始到第6月终点之间发生至少一次被指示为重度和/或以血浆葡萄糖≤3.9mmol/L(70mg/dL)所确认的夜间低血糖[00:00至05:59]的患者数目(%)(使用LOCF程序),-mITT群体
n(%)=发生至少一次被指示为重度和/或以血浆葡萄糖≤3.9mmol/L(70mg/dL)所确认的夜间低血糖的患者数目和百分比
a根据筛选HbA1c的随机化分层(<8.0或≥8.0%)分层的RR,使用CMH方法
2.2.2.2注射前血浆葡萄糖–第6月终点
表28–第二主要的次级功效终点–使用LOCF程序从基线至第6月终点的平均注射前SMPG(mmol/L)的平均值变化-mITT群体
LOCF=末次观测值结转。
SMPG=自我检测血浆葡萄糖
a平均值通过在给定访视之前7天内计算的至少3个SMPG的平均值进行评估。
b协方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层HbA1c(<8.0,≥8.0%)、和国家为固定效应,以基线平均注射前SMPG值作为协变量
注:对于所有在6个月期间接受救援的患者,使用救援前和6个月治疗期的最后一次基线后平均注射前SMPG测量值作为平均注射前SMPG终点
表28数据在图6中被总结。
2.2.2.3注射前SMPG的变异性–第6月终点
表29–第三主要的次级功效终点–使用LOCF程序从基线至第6月终点的注射前SMPG的平均值变化-mITT群体
LOCF=末次观测值结转
SMPG=自我监测血浆葡萄糖
变异性通过在给定访视之前7天内测量的至少3个SMPG计算的平均变异系数进行评估。
a协方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层HbA1c(<8.0,≥8.0%)、和国家为固定效应
注:对于所有在6个月期间接受救援的患者,使用救援前和6个月治疗期的最后一次基线后注射前SMPG变异性测量值作为注射前SMPG变异性终点
2.2.3其它次级功效终点
2.2.3.1在第6月时HbA1c<7%的患者百分比
表30–其它次级功效终点–在第6月终点HbA1c<7%的患者数目(%)(使用LOCF程序)和在第6月终点HbA1c<7%且在6个月主治疗期期间的最后3个月内没有经历被指示为重度和/或被血浆葡萄糖<3mmol/L(54mg/dL)所确认的低血糖的患者的数目(%)(使用LOCF程序)–mITT群体
LOCF=末次观测值结转.
RR=相对危险度
a根据筛选HbA1c的随机化分层(<8.0或≥8.0%)分层的RR,使用CMH方法
注:对于所有在6个月期间接受救援的患者,使用救援前和6个月治疗期的最后一次基线后HbA1c测量值作为HbA1c终点
2.2.3.2 FPG从基线至第6月的变化
表31-其它次级功效终点-使用LOCF程序从基线至第6月终点的FPG(mmol/L)平均值变化-mITT群体
FPG=空腹血浆葡萄糖
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型,以治疗组(HOE901-U300和LANTUS)、筛选HbA1c的随机化分层HbA1c(<8.0,≥8.0%)、和国家为固定效应,以基线FPG值作为协变量
注:对于所有在6个月期间接受救援的患者,使用救援前和6个月治疗期的最后一次基线后FPG测量值作为FPG终点
2.2.3.3八点SMPG概貌
在基线和第6月终点的平均八点SMPG概貌(mmol/l)在图7中有描述(mITT群体)。
2.2.3.4基础胰岛素剂量
在6个月主治疗期期间各次访视的平均每日基础胰岛素剂量(U)(mITT群体)在图8中描述。
2.3安全性评估
2.3.1暴露程度
表32–在6个月主治疗期期间对研究产品的暴露-安全性群体
注:患者视为在其随机化时实际接受的治疗组中
2.3.2低血糖
表33-在6个月主治疗期期间发生至少一次突发低血糖事件的患者数目(%)–安全性群体
n(%)=发生至少一次低血糖事件的患者数目和百分比
a重度和/或被确认的低血糖=重度和/或被血浆葡萄糖≤3.9mmol/L(70mg/dL)(分别≤3.0mmol/L(54mg/dL))所确认
表34-按研究期区分的在6个月主治疗期期间发生至少一次突发低血糖事件的患者数目(%)-安全性群体
n(%)=发生至少一次低血糖事件的患者数目和百分比
a重度和/或被确认的低血糖=重度和/或被血浆葡萄糖≤3.9mmol/L(70mg/dL)(分别≤3.0mmol/L(54mg/dL))所确认
2.3.3治疗引发的不良事件
表35–在6个月主治疗期期间由治疗引发的不良事件-安全性群体
TEAE:治疗引发的不良事件,SAE:严重不良事件
n(%)=发生至少一次TEAE的患者数目和百分比
表36–根据主要SOC,HLT和PT区分的在6个月主治疗期期间在任何治疗组中发生HLT≥2%的TEAE的患者数目(%)-安全性群体
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLT:高级别术语,PT:优选术语
MedDRA 16.0
n(%)=发生至少一次TEAE的患者数目和百分比
注:表格根据SOC国际公认次序排序,HLT,PT按照字母次序排序
只提供了在至少一个组中具有最少一个HLT≥2%的HLT
2.3.4死亡、严重的治疗引发的不良事件
2.3.4.1死亡
表37–在研究期间(研究中、治疗中、研究后)死亡的患者数目(%)-安全性群体
TEAE:治疗引发的不良事件,SAE:严重不良事件
a包括从治疗开始到研究结束(由末次方案计划访视或者所有治疗引发SAE的解决/稳定和预定的监视不良事件定义)之后发生的全部死亡
b治疗中是6个月主治疗期
c包括在研究结束之后(按照脚标a定义的)发生的并且被报告在数据库中的死亡
2.3.4.2严重不良事件
表38–按照主要SOC,HLGT,HLT和PT区分的在6个月主治疗期期间发生治疗引发SAE的患者数目(%)-安全性群体
SAE:严重不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语
MedDRA 16.0
n(%)=发生至少一次治疗引发SAE的患者数目和百分比
注:表格根据SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
2.3.5导致退出的不良事件
表39–按照主要SOC,HLGT,HLT和PT区分的在6个月主治疗期期间发生导致永久性治疗中断的TEAE的患者数目(%)-安全性群体
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语
MedDRA 16.0
n(%)=发生至少一次导致永久性治疗中断的TEAE的患者数目和百分比
注:表格根据SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
2.3.6其它显著的不良事件
2.3.6.1超敏反应
表40–根据相关标准化MedDRA问卷和优选术语-的超敏反应区分的在6个月主治疗期期间经历了至少一次TEAE的患者数目(%)–安全性群体
MedDRA 16.0
TEAE:治疗引发的不良事件
n(%)=发生至少一次超敏反应事件的患者数目和百分比
2.3.6.2注射部位反应
表41–根据相关标准化MedDRA问卷和优选术语-的注射部位反应区分的在6个月主治疗期期间经历了至少一次TEAE的患者数目(%)–安全性群体
MedDRA 16.0
TEAE:治疗引发的不良事件
n(%)=在注射部位事件中具有至少一次局部耐受性的患者的数目和百分比
2.3.7体重
表42–生命体征-描述性统计-使用LOCF程序在6个月主治疗期期间从基线至第6月终点的体重平均值变化(kg)–安全性群体
LOCF=末次观测值结转.
实施例3:在2型糖尿病患者中比较甘精胰岛素新制剂与的功效和安全性的6个月、多中心、随机、开放标签、平行分组研究,两组均添加餐时胰岛素,并具有6个月安全性延长期——比较可调适时间间隔和固定施用间隔的施用子研究
1.概要
开发阶段:3
目的:
主要目的:以从主研究的第6个月(=子研究的基线)到主研究的第9个月(=子研究的终点)的HbA1c变化为量度,比较每天每24小时注射一次HOE901-U300和每天以24±3小时的间隔注射一次HOE901-U300在2型糖尿病患者中的功效。
主要的次级目的:以夜间低血糖的发生率为量度,比较HOE901-U300的两种注射方案的安全性。
方法学:将随机分组使用HOE901-U300并且在6个月主要研究期间接受了HOE901-U300的患者以1:1随机分组为每天以每24小时(固定施用间隔)或每24±3小时(可调适时间间隔)施用一次HOE901-U300。
使用HOE901-U300完成了6个月主要研究期(见实施例1)并且满足子研究合格性标准的患者对该子研究适格。描述性的主要分析不要求具体的样本量。
患者数目:计划:最多达300名(每个治疗臂150名)
随机化:110
治疗:110
评估: 功效:109 安全性:110
诊断和纳入标准:
纳入标准:完成了实施例1所述主要研究(第10次访视)的6个月研究期,随机分组并在6个月治疗期间(基线-6个月)用HOE901-U300治疗,获得了子研究的签署书面知情同意书。
关键排除标准:患者不愿意每周在至少2天采用24±3小时的可调适的注射间隔;根据研究者的意见,不能够遵从可调适的给药时刻表;导致患者不能进一步参加本研究的健康状况。
研究治疗
研究性药品:测试药物:HOE901-U300
制剂:HOE901-U300(甘精胰岛素300U/mL溶液)是无菌、无热原、澄清、无色溶液,置于已经组装在笔式注射器的玻璃针筒中(预装填的,即一次性的笔)。
施用途径:皮下注射
测试方案:
可调适给药间隔:每天以24±3小时的间隔施用一次HOE901-U300。
注射时间可以根据个人的需要调适,比在主要研究开始时固定在夜间的每日参考注射时间早或晚最多3个小时。每周有至少2天使用最大间隔,即比固定的每日参考注射时间早3个小时或者晚3个小时,依患者选择。在主要研究开始时确定的注射时间保持作为变化的参考时间。
对照方案:
固定的给药间隔:HOE901-U300,每天每24小时注射一次。
患者继续每天每24小时在主要研究开始时确定的注射时间注射一次HOE901-U300。
剂量:
HOE901-U300的剂量根据需要进行滴定,以使空腹血浆葡萄糖达到或稳定在80~100mg/dL(4.4mmol/L~5.6mmol/L)的目标范围而没有低血糖。胰岛素剂量的改变以空腹自我监测的毛细血管血浆葡萄糖(SMPG)测量结果为根据。
非研究性药品:
两个治疗组的患者在该子研究期间继续使用其餐时胰岛素类似物。
同时使用二甲双胍治疗的患者在子研究期间继续使用稳定的剂量,除非因安全性问题而必须减少二甲双胍的剂量或中断二甲双胍。
治疗的持续时间:子研究的组成为3个月的比较功效和安全性治疗期,从6个月主要研究期完成开始至9个月主要研究完成时结束。
完成后,属于可调适给药臂的患者可能已持续该方案直至主研究结束(第12月)。每24小时注射一次HOE901-U300的患者持续其治疗方案直至主研究结束。
观察的持续时间:功效和安全性的分析期是3个月研究期,从主研究的第6月开始到主研究的第9月结束。本KRM中提供的结果是指这个时期。
评估标准:
功效:
主要功效终点:HbA1c从基线(第6月)到终点(第9月)的变化。
次级功效终点:FPG(中心实验)从基线(第6月)到终点(第9月)之间的变化,基础胰岛素和餐时胰岛素的每日剂量。
安全性:低血糖,不良事件特别是治疗引发的不良事件(TEAE)和严重不良事件(SAE)的发生,注射部位反应和超敏反应。下列信息没有在KRM中提供:其它安全信息,包括生命体征和用药过量。
统计方法:
对于本3个月子研究,基线定位为主研究的第6月;终点是主研究的第9月。
主要功效终点(从基线[第6月]到终点[第9月]的HbA1c变化)的分析使用协方差分析(ANCOVA)模型,以治疗方案和国家作为固定效应,以HbA1c的基线值作为协变量。HOE901-U300可调适的给药方案和HOE901-U300固定给药方案之间的差异和双侧95%置信区间在ANCOVA的框架内估算。
所有连续的次级功效变量(除注射前SMPG变异性的变化之外)使用ANCOVA模型分析,以治疗方案和国家作为固定效应,以相应的基线值作为协变量。
从基线(第6月)到终点(第9月)的注射前SMPG变异性的变化使用协方差分析(ANOVA)模型进行分析,以治疗方案和国家作为固定效应。
安全性分析是描述性的,基于安全性群体。
概要:
群体表征:
总共110名2型糖尿病患者被随机分组到子研究中:56名患者分组到HOE901-U300可调适给药间隔方案,54名患者分组到HOE901-U300固定给药间隔方案;110名患者暴露于IMP(安全性群体)。mITT子研究群体(功效群体)包括109名患者。
1名随机分组到HOE901-U300可调适给药间隔的患者(1/56,1.8%)提前中断了子研究,也中断了主研究的延长期(HOE901-U300固定给药间隔:0/54,0%)。
基线的人口学特征和患者特征在两个方案组之间良好平衡。子研究群体的平均年龄是60岁;110名患者中有35位(31.8%)≥65岁。
在子研究期间,可调适给药间隔组中的患者有平均23.0%的注射以距前次注射<21.5小时或>26.5小时的极端间隔实施,而固定给药间隔组中的患者为3.9%。可调适给药间隔组中的患者有平均13.5%的注射以中间间隔(前次注射后21.5-23小时或25-26.5小时)实施,而固定给药间隔组中的患者为8.2%。与固定给药间隔组(88.0%)相比,可调适给药间隔组有更少的患者(63.4%)在前次注射后23-25小时内注射。
可调适给药间隔组中有总共34.5%的患者在距前次注射23-25小时间隔外进行的注射少于20%,因此大约65%的患者被认为遵从可调适时间间隔方案。在固定给药间隔组中,78.8%的患者在距前次注射23-25小时间隔内进行了超过80%的注射,因此可以被认为遵从固定给药间隔方案。
当按照主研究基线时规划的参考注射时间计算间隔时,两个方案的顺应性相似。
功效结果:
主要终点:HbA1c从基线(第6月)到终点(第9月)的LS平均值变化在可调适给药间隔(0.22%[95%CI:-0.006至0.436])和固定给药间隔(0.14%[95%CI:-0.099至0.380])组之间相似。
次级功效终点(第9月):
·FPG(可调适给药间隔1.40mmol/L[95%CI:0.624至2.177];固定给药间隔(1.18mmol/L[95%CI:0.350至2.015];LS平均值变化0.22mmol/L[95%CI:-0.634至1.070])。
·注射前SMPG、注射前SMPG的变异性:
对HOE901-U300的注射间隔的调适导致间隔比规则的24小时间隔更短或更长,可能会影响注射前SMPG和注射前SMPG的变异性等次级功效终点。因此,除了总体分析之外,CSR中还会提供按照各注射间隔分解的SMPG数据。总体分析显示每个剂量间隔方案的患者在访视前最长达7天的平均均值。
在两个给药间隔方案组中,平均基础胰岛素和餐时胰岛素每日剂量在3个月比较方案期间保持稳定。
安全性:
在3个月比较方案期间,HOE901-U300可调适给药间隔方案和HOE901-U300固定给药间隔方案中有相似百分比的患者报告了低血糖事件,同时包括总体和每种分类的低血糖。
发生任何TEAE的患者(可调适给药间隔15/56[26.8%];固定给药间隔15/54[27.8%])或严重TEAE(可调适给药间隔4/56[7.1%];固定给药间隔5/56[9.3%])的百分比在两个方案之间相当。
在3个月子研究期间,每个给药间隔方案均未观察到导致治疗中断或导致死亡,或者与注射部位反应相关联的TEAE。固定给药间隔方案中有1名患者[1.9%]发生了与超敏反应有关的TAEA。
结论:
两个给药间隔方案组中大部分患者遵从了如随机化确定的注射时间表,并使用可调适给药间隔(每天一次HOE901-U300,每周有至少2天间隔每24小时±3小时)或固定给药间隔(每天一次HOE901-U300,每24小时)。这允许比较这两种给药间隔方案以进行功效和安全性分析。
以HbA1c和FPG为量度的功效分析显示两种给药间隔方案的结果相当。
两种方案在3个月子研究期间的低血糖总发生率(发生至少一次事件的患者%)相似,无论低血糖分类如何。
HOE901-U300可调适给药间隔和HOE901-U300固定给药间隔在3个月的比较子研究期内得到了良好耐受;没有观察到特定的安全性问题。
总之,根据子研究结果,未发现偶尔修改注射间隔对主要功效和安全性终点的负面作用。
2结果
2.1研究患者
2.1.1研究处置
表43-患者处置–随机子研究群体
注:百分数的计算使用随机化患者的数目作为分母
表44-子研究分析群体
注:患者根据其随机化的治疗列表
2.1.2人口学特征和基线特征
表45-基线处的人口学特征和患者表征-随机化子研究群体
年龄在主研究基线评估
表46-与注射时间相关的基线(第6月)功效数据-随机化子研究群体
平均值:在第6月之前的最后7天内至少3个时间间隔的平均间隔值
2.1.3治疗顺应性的测量
表47-顺应性–3个月比较方案期间的给药方案顺应性–根据给药间隔分类区分的注射百分比(距前次注射的时间)-安全性子研究群体
注:使用在第7.5月和第9月的访视之前最后7天中获取的全部间隔数值为每位患者计算每个给药间隔分类中注射的百分比(距前次注射的时间)
注:本表格中仅考虑了在第7.5月或第9月访视之前最后7天内数值具有至少3个时间间隔的患者。
表48-顺应性–在距离前次注射时间23至25小时的时间窗口之外的注射少于20%的患者的数目-安全性子研究群体
注:使用在第7.5月和第9月的访视之前最后7天中获取的全部间隔数值为每位患者计算每个给药间隔分类中注射的百分比(距前次注射的时间)。
注:本表格中仅考虑在第7.5月和第9月的访视之前最后7天内具有至少3个时间间隔数值的患者
表49-顺应性–在3个月比较方案期间的给药方案顺应性-按照给药间隔分类的注射百分数(距前次注射的时间)区分-安全性子研究群体
注:使用在第7.5月和第9月的访视之前最后7天中获取的全部间隔数值为每位患者计算每个给药间隔分类中注射的百分比(距前次注射的时间)。
注:本表格中仅考虑在第7.5月和第9月的访视之前最后7天内具有至少3个时间间隔数值的患者
2.2功效评估
2.2.1主要功效终点
表50-主要功效分析-使用LOCF程序从基线到第9月终点的HbA1c(%)平均值变化-mITT子研究群体
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型以治疗方案和国家作为固定效应,以基线HbA1c值为协变量
图9中描述了在3个月比较方案期间各次访视的平均HbA1c(%)-mITT子研究群体。
2.2.2次级终点
2.2.2.1空腹血浆葡萄糖
表51-使用LOCF程序从基线(第6月)到第9月终点的FPG平均值变化(mmol/L)-mITT子研究群体
FPG=空腹血浆葡萄糖.
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型以治疗方案和国家作为固定效应,以基线FPG值为协变量.
2.2.2.2基础和餐时胰岛素剂量
图10中描述了在3个月比较方案期间各次访视的平均每日基础(甘精胰岛素)和餐时胰岛素剂量(U)-mITT子研究群体。
2.3安全性评估
2.3.1暴露程度
表52-在3个月比较方案期间暴露于研究产品–安全性子研究群体
2.3.2低血糖
表53-在3个月比较方案期间发生至少一次低血糖的患者数目(%)-安全性子研究群体
n(%)=发生至少一次低血糖事件的患者数目和百分比
a重度和/或被确认的低血糖=重度和/或被血浆葡萄糖<=3.9mmol/L(70mg/dL)(分别<3.0mmol/L(54mg/dL))所确认
注:所有缺失时间的低血糖事件被计数在“全部低血糖”列中,但没有被分类成“夜间”或“日间”
2.3.3治疗引发的不良事件
表54-3个月比较方案期间的治疗引发的不良事件-安全性子研究群体
TEAE:治疗引发的不良事件,SAE:严重不良事件.
n(%)=发生至少一次TEAE的患者数目和百分比
表55-根据主要SOC,HLGT,HLT和PT事件区分的在3个月比较方案期间发生TEAE的患者数目(%)-安全性子研究群体
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语.
MedDRA 16.0.
n(%)=发生至少一次TEAE的患者数目和百分比
注:表格是按照SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
2.3.4严重的治疗引发不良事件
表56-根据主要SOC,HLGT,HLT和PT区分的在3个月比较方案期间发生治疗引发SAE的患者的数目(%)-安全性子研究群体
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语.
MedDRA 16.0.
n(%)=发生至少一次治疗引发SAE的患者数目和百分比
注:表格是按照SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
2.3.5导致退出的治疗引发的不良事件
表57-根据主要SOC,HLGT,HLT和PT区分的在3个月比较方案期间发生了导致永久性治疗中断的TEAE的患者数目(%)-安全性子研究群体
2.3.6其它显著的治疗引发的不良事件
2.3.6.1注射部位反应
表58-按照相关标准化MedDRA问卷和优选术语-注射部位反应区分的3个月比较方案期间经历了至少一次TEAE的患者数目(%)-安全性子研究群体
2.3.6.2超敏反应
表59-按照相关标准化MedDRA问卷和优选术语-超敏反应区分的3个月比较方案期间经历了至少一次TEAE的患者数目(%)–安全性子研究群体
TEAE:治疗引发的不良事件,PT:优选术语.
MedDRA 16.0
n(%)=发生了至少一次超敏反应TEAE的患者数目和百分比
注:表格根据HOE901-U300可调适给药间隔方案中PT的递减频率排序
实施例4:在1型糖尿病患者中比较甘精胰岛素新制剂与的功效和安全性的16周、随机化、开放标签、对照研究
开发阶段:2
目的:
主要目的:在患有1型糖尿病的成年患者中比较用甘精胰岛素新型制剂(HOE901-U300)和Lantus的葡萄糖控制。
次级目的:
·比较晨间或夜间给予的HOE901-U300和Lantus的连续葡萄糖监测(CGM)数据:日间葡萄糖暴露;以中位数曲线变化率度量的日间葡萄糖稳定性;以四分位距(IQR)测量的日间葡萄糖变异性;葡萄糖概貌的平均值和变异。
·比较HOE901-U300和Lantus的糖化血红蛋白A1c(HbA1c)、自我测量的血浆葡萄糖(空腹血浆葡萄糖,注射研究药物之前,7-点概貌)。
·比较下述分类的低血糖发作的发生率和频率:如ADA标准定义的;均有症状的;通过血浆葡萄糖>70mg/dL确认的;和CGM检测到的。
·评估HOE901-U300的安全性和耐受性。
方法学:在1糖尿病患者中比较HOE901-U300和Lantus的多中心、开放标签、随机化、4臂平行分组、比较性2期研究。使患者随机接受:每日一次基础胰岛素(HOE901-U300或Lantus);和在研究期A和研究期B内的注射时间顺序(晨间然后夜间,或者夜间然后晨间),比例为1:1:1:1。此探索性研究未进行正式的样品量估计。
患者数目:计划56名,随机化59名,治疗59名,评估:功效59名,安全性59名。
诊断和纳入标准:纳入标准:1型糖尿病患者;签署了书面知情同意书。关键排除标准:年龄<18岁和>70岁;筛选时HbA1c>9%;使用基础+餐时胰岛素短于1年;患者在筛选访视前的30天内接受>0.5U/kg体重的基础胰岛素,以及患者没有接受稳定胰岛素剂量(±20%总基础胰岛素剂量);在随机之前6个月内由于糖尿病酮症酸中毒住院或具有严重低血糖历史(需要第三方协助)。
研究治疗
研究性药品:测试药物:HOE901-U300;对照药物:Lantus
制剂:HOE901-U300作为装在3mL针筒中的300U/mL皮下(SC)注射用甘精胰岛素溶液提供。Lantus作为装在10mL小瓶中的SC注射用100U/mL甘精胰岛素溶液提供。
施用途径:SC注射
HOE901-U300注射是通过带半单位刻度(half-unit-scale)的市售BD Ultra-FineTM短针头胰岛素注射器进行。Lantus的注射用市售的BD胰岛素注射器进行:
·对于1–30U甘精胰岛素剂量:BD Ultra-FineTM短针头胰岛素注射器,带半单位刻度[8mm(5/16”)x 31G];
·对于>30U甘精胰岛素的剂量:BD Ultra-FineTM短针头胰岛素注射器,带半单位刻度[8mm(5/16”)x 31G]。
施用方案:根据随机分组,在周期A中每天在晨间或夜间注射一次,历时8周,然后在周期B中每天在夜间或晨间注射一次,历时8周。
起始剂量:在基线访视之前使用Lantus或者每天一次NPH或每天一次地特胰岛素的患者:HOE901-U300或Lantus的每日剂量(U)等于基线访视之前一天的基础胰岛素日剂量。在基线访视之前使用超过每天一次NPH或地特胰岛素的患者:使用基线访视前一天NPH或地特胰岛素每日总剂量的80%(=每日总剂量减少20%)。
研究期间的剂量:作为HOE901-U300或Lantus给予的甘精胰岛素施用是根据自我测量的空腹早餐前血浆葡萄糖水平(目标范围80–130mg/dL;4.4–7.2mmol/L)并考虑低血糖的存在而进行的。基础胰岛素的最小剂量增加是1.5U。
批次号:HOE901-U300:C1011129;Lantus:由当地药房提供。
非研究性药品:短效餐时(餐前)胰岛素类似物(谷赖、门冬或赖脯胰岛素)。
两个治疗组的患者在研究期间均继续使用其餐时胰岛素类似物。
餐时胰岛素剂量根据SMPG数据调整,包括2-小时餐后血浆葡萄糖结果和餐食糖类含量,以优化血糖控制。2小时餐后血浆葡萄糖的目标范围是<160mg/dL(8.3mmol/L)。餐前胰岛素剂量可以随着基础胰岛素剂量增加而降低。
治疗持续时间:最多达16周(周期A期间8周,周期B期间8周)。
观察持续时间:
·最多达4周筛选(包括2周CGM训练期);
·2x8周比较功效和安全性治疗期;
·研究完成或永久性中断之后的4周治疗后安全性跟踪期。
总计最大研究持续时间为每位患者多达24周。
评估标准:
功效
主要功效终点:根据CGM在治疗期A的第7和8周期间和治疗期B的第15和16周期间血糖范围在80-140mg/dL(4.4-7.8mmol/L)的时间的百分比(%)。
次级功效终点:高于血糖范围上限/低于下限的百分比时间(处于高血糖/低血糖的%时间)。
本KRM中没有提供下面的次级功效终点:日间葡萄糖暴露;日间葡萄糖稳定性;日间葡萄糖变异性;葡萄糖概貌的平均值和变异;在8周治疗期的最后2周中使用CGM的14天里,在每次给药间隔的后4个小时里处于血糖范围的平均时间;高血糖AUC(位于CGM概貌的下方、并高于血糖范围除以总时期的上限的面积);和低血糖AUC(位于CGM概貌的上方并低于血糖范围除以总时间周期的下限的面积)。
进一步的次级功效终点:胰岛素剂量;本KRM中没有提供:HbA1c,空腹血浆葡萄糖(FPG),注射前SMPG,7-点SMPG。
安全性
低血糖、不良事件特别是治疗引发的不良事件(TEAE)和严重不良事件(SAE)的发生率,注射部位反应和超敏反应。下面的信息没有在KRM中提供:身体检查,其它安全性信息包括临床实验数据、生命体征和12-导联ECG。
统计方法:
主要终点使用线性混合模型进行分析,以治疗(HOE901-U300或Lantus)和时期(治疗期A或B)作为固定效应,患者作为随机效应。提供了每个治疗的调整平均值估计和标准误,治疗平均差异的调整估计和标准误以及治疗平均差异的95%置信区间。统计检验是双侧检验,名义显著水平为5%。同一模型还用于次级功效终点高血糖/低血糖的%时间、日间葡萄糖暴露、日间葡萄糖稳定性、和日间葡萄糖变异性。其它功效终点是描述性的。CGM相关参数根据CGM群体进行分析,非CGM参数根据mITT群体分析。
安全性分析是描述性的,基于安全性群体。
概要:
群体表征:
总共59名1型糖尿病患者随机分到4个臂之一:HOE901-U300在周期A中在晨间注射然后在周期B中夜间注射(n=15),HOE901-U300在夜间然后在晨间注射(n=15),Lantus在晨间然后在夜间注射(n=15),或Lantus在夜间然后在晨间注射(n=14)。总共59名患者暴露于IMP(安全性群体,并且包含在mITT和CGM群体(功效群体)中。HOE901-U300组中有1名患者(3.3%)和Lantus组中有3名患者(10.3%)提前中断了研究治疗。
人口学特征和基线特征在治疗组之间平衡。研究群体的平均年龄为44.2岁,2名患者为65岁或更大。所有患者均是高加索人。基线时的平均BMI为27.3kg/m2。研究开始之前的糖尿病平均持续时间是22.1年。每日总胰岛素的中位数剂量是0.565U/kg体重。基线时的平均HbA1c是7.46%。
功效结果:
主要功效终点:在每个8周治疗期的最后2周里,在此时期基础胰岛素剂量要保持尽可能稳定,观察通过CGM测量的血浆葡萄糖发现,HOE901-U300组中有31.75%(LS平均值)的时间在血糖范围内,Lantus组中则是30.99%(LS平均值)的时间。LS均值差异是0.75%[95%CI:-3.614-5.124]。
次级功效终点:在每个8周治疗期的最后2周里,超过血糖范围上限140mg/dL(7.8mmol/L)的百分比时间在各治疗组之间相当(HOE901-U300组是58.24%,Lantus是57.38%,按照LS平均值计),低于下限80mg/dL(4.4mmol/L)的百分比时间也是如此,HOE901-U300组是10.01%,Lantus是11.64%,按照LS平均值计。
根据整个研究期间每天各小时点的CGM的平均葡萄糖的图示表明(图11),HOE901-U300组相比Lantus组漂移(excursion)较小。HOE901-U300的概貌与Lantus相比显得较为平坦,在晨间注射期(图12)中比夜间注射期(图13)中更加明显。
总之,在HOE901-U300和Lantus治疗组中,基础胰岛素类似地大多在研究的前6周增加,之后保持相对稳定(基线时,平均每日基础胰岛素剂量在两个治疗组中相似:HOE901-U300:24.9单位;Lantus:25.0单位;在第16周,HOE901-U300:30.11单位;Lantus:28.22单位)。
基线时,HOE901-U300组的平均餐时胰岛素每日剂量(29.92单位)高于Lantus组(23.69单位),但是在第16周时二者相当(HOE901-U300:27.34单位;Lantus:26.31单位)。
安全性结果:
在治疗期期间,就总体低血糖事件和每种分类的低血糖事件(全部低血糖)而言,HOE901-U300组和Lantus组中经历了低血糖的患者百分数相当。与之一致地,在下列亚组之间观察到了相似的低血糖报告:
·HOE901-U300组中的晨间和夜间注射组;
·Lantus组中的晨间和夜间注射组;
·HOE901-U300晨间注射组和Lantus晨间注射组;
·HOE901-U300夜间注射组和Lantus夜间注射组;
经历夜间低血糖的患者百分比在HOE901-U300组始终低于Lantus组,无论晨间还是夜间注射时间。由于患者的数目小,对HOE901-U300组的这种有利数值趋势的解释必须谨慎。
发生任何TEAE的患者的百分比在HOE901-U300组(24/30[80.0%])高于Lantus组(19/29[65.5%])。在HOE901-U300组中,1名患者经历了严重肠梗阻(与IMP无关),另1名患者由于怀孕中断了治疗。在研究期间没有报告死亡。在HOE901-U300组中有2/30[6.7%]名患者、在Lantus组中有1/29[3.4%]名患者观察到与注射部位反应关联的TEAE。不存在与超敏反应关联的TEAE的担忧,HOE901-U300组有4/40名患者出现超敏反应,Lantus组中有1/30患者出现超敏反应。
结论:
在每个8周治疗期的最后2周里,通过CGM测量的血浆葡萄糖处于目标范围(80-140mg/dL或4.4-7.8mmol/L)内的时间的百分比在HOE901-U300组与Lantus组之间相似。注意,这个目标范围比ADA推荐的70-180mg/dL(3.9-10.0mmol/L)更加紧凑。
在两个治疗组中,在血糖范围上限上方经历的时间的百分比(57%-58%)比在目标范围下限下方经历的时间的百分比(10-11%)高。
总体上,在研究期间发生至少一次事件(无论低血糖分类为何)的患者的百分比在两个治疗组(HOE901-U300,Lantus)之间和对于两种注射时间(晨间或夜间)均相当。由于患者的数目小,对HOE901-U300组对于夜间低血糖的有利数值趋势的解释必须谨慎。
HOE901-U300和Lantus在晨间或夜间施用在研究期内均得到了良好耐受,没有观察到特定的安全性问题。
实施例5:研究性新胰岛素U300:在基础胰岛素用药的2型糖尿病中的葡萄糖控制和低血糖(第II版)
目的:一种研究性新胰岛素U300,其具有比甘精胰岛素100U/mL(U100)更平坦和更长期的PK/PD概貌,正在临床开发中。该3期第II版研究比较了U300和U100在使用基础胰岛素方案并联用OAD的T2DM人中的功效和安全性。
方法:在此次多中心、开放标签、6个月的研究中,参加者被随机(1:1)分配给予每天夜间一次U300或U100。胰岛素剂量经过滴定,以达到80-100mg/dL的目标空腹血浆葡萄糖(FPG)。主要终点是从基线到第6月的HbA1c变化,分层分析中第一主要的次级功效终点是从第9周到第6月发生≥1次重度或被确认的(≤70mg/dL)夜间(2400–0559h)低血糖事件的患者百分比。
结果:811名参加者被随机分组[平均年龄58.2(SD 9.2)岁,糖尿病持续时间12.6(7.0)年,BMI 34.8(6.4)kg/m2,基础胰岛素剂量0.67(0.24)U/kg]。各组的基线HbA1c相当;U300:8.26(0.86)%对U100:8.22(0.77)%。对于HbA1c变化而言,U300不劣于U100[6个月时LS平均值变化分别是-0.57(SE:0.09)%和-0.56(SE:0.09)%;差异-0.01(95%CI:-0.14至+0.12)%]。对于FPG、8-点自我监测血浆葡萄糖概貌和注射前血浆葡萄糖,没有见到相关差异。从第9周到6个月发生重度或确认的夜间低血糖的患者百分比U300比U100显著低[21.6%对27.9%;相对危险度(RR)0.77(95%CI:0.61至0.99);p=0.038]。在整个6个月治疗期期间,任何夜间低血糖的发生率(发生≥1次事件的患者的%)U300比U100低[30.5%对41.6%;RR 0.73(95%CI:0.60至0.89)],每天任何时刻(24h)任何低血糖事件的发生率也是如此[U300 71.5%;U100 79.3%;RR 0.90(95%CI:0.84至0.97)]。U300中有1.0%而U100参与者中有1.5%报告了每天任何时刻的重度低血糖。未见治疗与治疗之间在严重不良事件方面存在的差异。
结论:在使用基础胰岛素方案和OAD的T2DM患者中,U300得到了良好耐受,并且在血糖控制方面与U100同样有效。从第9周到第6月,U300中重度或被确认的夜间低血糖比U100减少23%,并且在整个6个月研究持续时间内,U300中任何夜间低血糖和每天任何时刻(24h)低血糖的发生率比U100低。
实施例6:在2型糖尿病患者中比较甘精胰岛素新制剂与Lantus的功效和安全性的6个月、多中心、随机化、开放标签、平行分组研究,两组均与口服抗高血糖药物联用,并具有6个月安全性延长期——比较可调适给药间隔和固定给药间隔的施用子研究
梗概
开发阶段:3期主研究的子研究
目的:
主要目的:在2型糖尿病患者中比较每天以每24小时间隔注射一次HOE901-U300和每天以24±3小时的间隔注射一次HOE901-U300的功效,以HbA1c从主研究的第6个月(=子研究的基线)到主研究的第9个月(=子研究的终点)的变化为量度。
主要的次级目的:以夜间低血糖的发生率为量度,比较HOE901-U300的两种注射方案的安全性。
方法学:
对于被随机分派到HOE901-U300并在6个月主研究期间接受了HOE901-U300的患者,以1:1随机分派每天以24小时间隔(固定给药间隔)施用一次、或24±3小时间隔(可调适给药间隔)施用一次。
分组到HOE901-U300、完成了6个月主研究期、并且满足子研究适格标准的患者对于本子研究是适格的。描述性的主要分析不要求特定的样本量。
患者数目:计划:最多300人(每个治疗臂150人)
随机化:89
治疗:87
评估:功效:86 安全性:87
诊断和纳入标准:
纳入标准:完成主研究的6个月研究期(第10访视),在6个月治疗期间(基线-第6月)随机分组并用HOE901-U300治疗,获得了子研究的签署书面知情同意书。
关键排除标准:患者不愿意每周在至少2天采用24±3小时的可调适的注射间隔;按研究者的意见,无法遵从可调适的施用时刻表;导致患者不能进一步参加本研究的健康状况。
研究治疗
研究性药品:测试药物:HOE901-U300
制剂:HOE901-U300(甘精胰岛素300U/mL溶液)是无菌、无热原、澄清、无色溶液,置于已经组装在笔式注射器的玻璃针筒中(预装填的,一次性的笔)。
施用途径:皮下注射
测试方案:
可调适给药间隔:每天以24±3小时的间隔施用一次HOE901-U300。
注射时间可以根据个人的需要调适到比每日夜间参考注射时间(该时间在主研究开始时固定)早或晚最多3个小时。根据患者的选择,每周有至少2天使用最大间隔,即比固定的每日参考注射时间早3个小时或者晚3个小时。在主研究开始时固定的注射时间可以保持,作为变异的参考时间。
对照方案
固定的给药间隔:HOE901-U300,每天每隔24小时注射一次。
患者继续每天每隔24小时在主研究开始时固定的注射时间注射一次HOE901-U300。
剂量:
根据需要滴定HOE901-U300的剂量,以使空腹血浆葡萄糖达到或者稳定在80-100mg/dL(4.4mmol/L-5.6mmol/L)的范围而不发生低血糖。胰岛素剂量的改变根据空腹自我监测的毛细血管血浆葡萄糖(SMPG)测量结果进行。
非研究性药品:
两个治疗方案的患者在参加本子研究期间继续使用其口服抗高血糖背景疗法。剂量在整个研究中保持稳定,除非发生与这些治疗相关的特定安全性问题。在本研究中不使用其他伴随的抗糖尿病治疗。
短期使用(即,最多10天)短效胰岛素疗法(例如,由于急性病或手术)不视为救援疗法。救援用药视为非研究性药品。
治疗的持续时间:子研究由3个月的比较功效和安全性治疗期组成,从6个月主研究期完成开始,至9个月主研究完成时结束。
完成后,属于可调适施用臂的患者可以继续执行该方案直至主研究结束(第12月)。每24小时注射一次HOE901-U300的患者继续执行其治疗方案直至主研究结束。
观察的持续时间:功效和安全性的分析期是从主研究的第6月开始到主研究的第9月结束的3个月研究期。本KRM中提供的结果是指这个时期。
评估标准:
主要功效终点:HbA1c从基线(第6月)到终点(第9月)的变化。
次级功效终点:FPG(中心实验)和每日基础胰岛素剂量从基线(第6月)到终点(第9月)之间的变化。
安全性:低血糖(包括夜间),不良事件特别是治疗引发的不良事件(TEAE)和严重不良事件(SAE)的发生率,导致退出的TEAE、注射部位反应和超敏反应。下列信息没有在KRM中提供:其它安全信息,包括生命体征和用药过量。
统计方法:
对于本3个月子研究,基线定义为主研究的第6月;终点是主研究的第9月,使用末次观测值结转(LOCF)程序。
主要功效终点(从基线[第6月]到终点[第9月]的HbA1c变化)的分析使用协方差分析(ANCOVA)模型,以治疗方案和国家作为固定效应,并使用HbA1c的基线值作为协变量。在ANCOVA的框架内估算HOE901-U300可调适的施用方案和HOE901-U300固定施用方案之间的差异以及双侧95%置信区间。
所有连续的次级功效变量均使用ANCOVA模型分析,以治疗方案和国家作为固定效应,使用相应的基线值作为协变量。
摘要
群体特征:
总共89名患有2型糖尿病的患者被随机分组到子研究:45名患者分组到HOE901-U300可调适给药间隔方案,44名患者分组到HOE901-U300固定给药间隔方案;87名患者暴露于IMP(安全性群体)。mITT子研究群体(功效群体)包括86名患者。
总共有40名(88.9%)随机分组到HOE901-U300可调适给药间隔的患者和38名(86.4%)随机分组到HOE901-U300固定给药间隔的患者完成了3个月比较方案期。1名(2.2%)随机分组到HOE901-U300可调适给药间隔的患者和2名(4.5%)随机分组到HOE901-U300固定给药间隔的患者提前中断了该子研究,也中断了主研究的延长期。
基线(第6月)人口学特征和患者特征在两个方案组之间得到了良好的平衡。子研究群体的均值年龄是57.8岁;89名患者中有16名(18.0%)≥65岁。可调适给药间隔方案的3名患者和固定给药间隔方案的2名患者在主研究期间启动胰岛素或胰岛素促分泌素(secretagogue)作为救援治疗。在3个月比较方案期间,两个方案组中均没有患者启动救援治疗。
对给药间隔方案的顺应性:
对给药间隔方案的顺应性评估考虑到了两次连续注射之间的时间间隔,以及注射与在主研究基线时设定的参考注射时间之间的时间间隔。
在子研究期间,可调适给药间隔组每位患者有平均28.04%的注射以两次连续注射之间<21.5小时或>26.5小时的极端间隔发生,而固定给药间隔组的患者有2.41%的注射以此间隔发生,同时,固定给药间隔组的每位患者有88.77%的注射以两次连续注射之间23-25小时的间隔发生,而可调适给药间隔组的患者有53.09%的注射以此间隔发生。
实际注射时间与参考注射时间之间的时间间隔评估显示,与可调适给药间隔组(56.38%)相比,固定给药间隔组的每位患者有更高百分比的注射发生在23-25小时间隔内(均值65.07%)。在可调适给药间隔组中,每位患者有21.69%的注射以21.5-23小时间隔或者以25-26.5小时间隔发生(固定给药间隔组为25.51%)。这些数据表明,大部分注射在参考注射时间之前或之后最多3小时内发生,参考参考注射时间按照规程被固定在夜间。
根据第7.5月或第9月访视之前一周中记录的注射时间,在可调适施用组中有总共47.5%的患者有4个或更多个注射间隔在距前次注射21.5-26.5小时的间隔之外,因此认为遵从了可调适给药间隔方案,而在固定给药间隔方案中的这样的患者有2.6%。在固定给药间隔组中,在61.5%的患者中所有连续注射间隔均在23-25小时之内,因此可以认为这些患者遵从了固定的给药间隔方案。
功效:
主要功效终点(第9月):从基线(第6月)到终点(第9月)的HbA1c LS平均值变化在可调适给药间隔组(-0.12%[95%CI:-0.422至0.183])和固定给药间隔组(-0.25%[95%CI:-0.574至0.072])之间相似,LS均值差异为0.13%[95%CI:-0.152至0.415]。
次级功效终点(第9月):从基线(第6月)到终点(第9月)的FPG LS平均值变化在可调适给药间隔组(-0.46mmol/L[95%CI:-1.521至0.609])和固定给药间隔组(-0.25mmol/L[95%CI:-1.378至0.881])之间相似,LS均值差异为-0.21[95%CI:-1.200至0.784]。
在两个给药间隔方案中,平均每日基础胰岛素剂量在3个月比较方案期间保持稳定。
安全性:
在3个月比较子研究期间,可调适给药间隔方案有16/44(36.4%)的患者报告低血糖事件,而固定给药间隔方案有18/43(41.9%)的患者报告低血糖事件。两个方案中报告每种分类低血糖事件的患者百分比相当。在任一方案中均未发生重度低血糖或重度夜间低血糖事件。
发生任何TEAE(可调适给药间隔9/44[20.5%];固定给药间隔11/43[25.6%])或严重TEAE(可调适给药间隔2/44[4.5%];固定给药间隔0/43)的患者百分比在各方案之间相当。
在3个月子研究期间,任一给药间隔方案均未观察到导致治疗中断或死亡、或者与注射部位反应或超敏反应相关联的TEAE。
结论:
注射间隔持续时间和注射间隔短于或长于规则24小时的%患者的评估表明:两个给药间隔方案组中的大部分患者遵从了随机分组的注射时刻表,并使用了可调适给药间隔(每天一次HOE901-U300,每周有至少2天间隔每隔24小时±3小时)或固定给药间隔(每天一次HOE901-U300,每隔24小时)。这允许比较两个给药间隔方案的功效和安全性分析。
以HbA1c和FPG从基线(第6月)到终点(第9月)的变化为量度的功效分析显示,两个给药间隔方案具有相当的结果。
在3个月子研究期间的低血糖总发生率(发生至少一次事件的患者%)在两个方案中相当,无论低血糖分类为何。
以可调适的给药间隔或固定的给药间隔给予的HOE901-U300在3个月比较研究期间得到良好耐受;没有观察到特定的安全性问题。
总之,子研究结果表明,与以24小时间隔每日注射一次相比,将每日注射一次HOE901-U300的注射时间偶尔调适为比参考时间早或晚最多3个小时,不会影响主要功效(HbA1c)和安全性终点,特别是低血糖事件。
2.结果
2.1研究患者
2.1.1研究处置
表60-患者处置–随机子研究群体
注:百分比计算使用被随机化的患者数目作为分母
a包括在主要6个月期间开始救援治疗并在3个月比较方案期间继续进行的患者
表61-子研究分析群体
注:患者根据其随机化的治疗制表
2.1.2人口学特征和基线特征
表62-基线时的人口学特征和患者表征-随机子研究群体
年龄在主研究基线时评估
表63-与给药间隔相关的基线(第6月)功效数据-随机化子研究群体
平均:在第6月之前的最后7天内在至少3个时间间隔上的平均间隔值
2.1.3对给药间隔方案的顺应性
表64-顺应性–3个月比较方案期间的剂量间隔方案顺应性–根据给药间隔分类(两次连续注射之间的时间)区分的每位患者的注射百分比-安全性子研究群体
注:使用在第7.5月和第9月访视之前最后7天获得的全部注射间隔数值为每位患者计算每个给药间隔分类中的注射百分比(两次连续注射之间的时间)
注:只有在第7.5月和第9月访视之前最后7天内具有至少3个时间间隔的患者才在本表中被考虑
表65-顺应性–3个月比较方案期间的剂量间隔方案顺应性–根据两次连续注射之间的注射间隔的患者数目(%)-安全性子研究群体
注:使用在第7.5月和第9月访视之前最后7天获得的全部注射间隔数值(最大12)计算注射数目(2个连续注射之间的时间)
表66-顺应性–3个月比较方案期间的剂量间隔方案顺应性–根据给药间隔分类的注射百分比(从参考注射开始的时间)-安全性子研究群体
注:使用在第7.5月和第9月访视之前最后7天获得的全部注射间隔数值为每位患者计算每个给药间隔分类中的注射百分比(从在主研究开始时选择参考注射时开始的时间)
注:只有在在第7.5月和第9月访视之前最后7天内具有至少3个时间间隔的患者才被考虑在本表中
2.2功效评估
2.2.1主要功效终点
表67–主要功效分析-使用LOCF程序从基线到第9月终点的HbA1c(%)均值变化-mITT子研究群体
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型,以治疗方案和国家作为固定效应,以基线HbA1c值为协变量
注:对于所有在3个月比较方案期间接受救援的患者,使用救援前和3个月治疗周期子研究期间的基线HbA1c最后一次测量值作为HbA1c终点
图14描述了在3个月比较方案期间访视的平均HbA1c(%)-mITT子研究群体.
2.2.2次级终点
2.2.2.1空腹血浆葡萄糖
表68-使用LOCF程序从基线到第9月终点的FPG平均值变化(mmol/L)-mITT子研究群体
FPG=空腹血浆葡萄糖.
LOCF=末次观测值结转.
a协方差分析(ANCOVA)模型,以治疗方案和国家作为固定效应,以基线FPG值为协变量
注:对于在3个月比较方案期间接受救援的所有患者,使用救援前和3个月治疗周期子研究期间的基线FPG最后一次测量值作为FPG终点
2.2.2.2基础胰岛素剂量
图15描述了在3个月比较方案期间各次访视的平均每天基础(甘精胰岛素)胰岛素剂量(U)–mITT子研究群体
2.3安全性评估
2.3.1暴露程度
表69-在3个月比较方案期间对研究制品的暴露–安全性子研究群体
2.3.2低血糖
表70-在3个月比较方案期间发生至少一次低血糖事件的患者数目(%)-安全性子研究群体
n(%)=发生至少一次低血糖事件的患者数目和百分比
a重度和/或被确认的低血糖=重度和/或被血浆葡萄糖≤3.9mmol/L(70mg/dL)(分别<3.0mmol/L(54mg/dL))所确认
注:缺失时间的所有低血糖事件被计数在“全部低血糖”列中,但是没有被分类为“夜间”或“日间”
2.3.3治疗引发的不良事件
表71-在3个月比较方案期间的治疗引发的不良事件-安全性子研究群体
TEAE:治疗引发的不良事件,SAE:严重不良事件.
n(%)=发生至少一次TEAE的患者数目和百分比
表72-按主要SOC,HLGT,HLT和PT事件区分的在3个月比较方案期间发生了TEAE的患者数目(%)
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语.
MedDRA 16.0.
n(%)=发生发生至少一次TEAE的患者数目和百分比
注:表格是按照SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
2.3.4严重治疗引发的不良事件
表73-按照主要SOC,HLGT,HLT和PT区分的在3个月比较方案期间发生了治疗引发SAE的患者数目(%)-安全性子研究群体
TEAE:治疗引发的不良事件,SOC:系统器官分类,HLGT:高级别组术语,HLT:高级别术语,PT:优选术语.
MedDRA 16.0.
n(%)=发生了至少一次治疗引发SAE的患者数目和百分比
注:表格是按照SOC国际公认次序排序,HLGT,HLT,PT按照字母次序排序
2.3.5导致退出的治疗引发的不良事件
表74-按照主要SOC,HLGT,HLT和PT区分的在3个月比较方案期间发生了导致永久性治疗中断的TEAE的患者数目(%)-安全性子研究群体
2.3.6其它显著治疗引发的不良事件
2.3.6.1注射部位反应
表75-按照相对标准化MedDRA问卷和优选术语-注射部位反应区分的在3个月比较方案期间经历了至少一次TEAE的患者数目(%)–安全性子研究群体
2.3.6.2超敏反应
表76-按照相对标准化MedDRA问卷和优选术语-超敏反应区分的在3个月比较方案期间经历了至少一次TEAE的患者数目(%)-安全性子研究群体
- 还没有人留言评论。精彩留言会获得点赞!