用他塞利昔布进行治疗的方法与流程
相关申请的交叉引用
根据37cfr§1.53(b)提交的这篇非临时申请根据35usc§119(e)要求2015年6月29日提交的美国临时申请系列no.62/186,236的权益,将该临时申请通过提述完整并入本文。
发明领域
本发明总体上涉及用pi3k抑制剂化合物他塞利昔布(taselisib)(gdc-0032)治疗癌症。本发明还涉及使用他塞利昔布进行体外,原位和体内诊断或治疗哺乳动物细胞或相关病理状况的方法。
发明背景
磷酸肌醇3-激酶(pi3k)/akt信号通路的上调是大多数癌症的常见特征(yuan和cantley(2008)oncogene27:5497-510)。在许多人类癌症中,已经检测到该通路的遗传偏离(osaka等人(2004)apoptosis9:667-76),所述遗传偏离主要起刺激细胞增殖,迁移和存活的作用。该通路的激活发生在激活编码p110αpi3k同等型的pik3ca基因的点突变或扩增之后(samuels等人(2004)science304:554;hennessy等人(2005)nat.rev.drugdiscov.4:988-1004)。肿瘤抑制蛋白pten(一种与pi3k具有相反功能的磷酸酶)的遗传缺失或功能丧失突变也增加了pi3k通路信号传导(zhang和yu(2010)clin.cancerres.16:4325-30)。
这些畸变通过诸如akt和mtor的激酶导致下游信号传导增加,人们已经提出pi3k通路的活性增加为癌症治疗抵抗的标志(opel等人(2007)cancerres.67:735-45;razis等人(2011)breastcancerres.treat.128:447-56)。
磷脂酰肌醇3-激酶(pi3k)信号传导通路是激素受体(hr)阳性转移性乳腺癌(mbc)中最失调的通路之一(bachmanke等人,cancerbiolther.2004;3:772-775;stemke-halek等人,cancerres.2008;68:6084-6091;koboldtdc等人,nature2012;490:61-70)。磷脂酰肌醇-4,5-二磷酸3-激酶催化亚基α(pik3ca)编码pi3k催化亚基的pi3kα同等型(p110)(samuelsy等人,science2004;304:554),在约40%的hr阳性bc中检出该基因的突变(arthurlm等人,breastcancerrestreat.2014;147:211)。
磷脂酰肌醇酯3-激酶(pi3k)是淋巴瘤的关键存活和生长信号的主要信号传导节点,与磷酸酶pten的活性相反。pi3k通路在侵袭性淋巴瘤中失调(abubaker(2007)leukemia21:2368-2370)。8%的dlbcl(弥漫性大b细胞淋巴瘤)癌症具有pi3kca(磷脂酰肌醇-3激酶催化亚基α)错义突变,37%的dlbcl的免疫组织化学试验为pten阴性。
磷脂酰肌醇是细胞膜中发现的多种磷脂之一,其参与细胞内信号转导。通过3′-磷酸化磷酸肌醇的细胞信号传导已经牵涉多种细胞过程,例如恶性转化,生长因子信号传导,炎症和免疫(rameh等人(1999)j.biolchem.274:8347-8350)。负责生成这些磷酸化信号传导产物的酶,即,磷脂酰肌醇3-激酶(也称为pi3激酶或pi3k)最初被鉴定为与病毒癌蛋白和生长因子受体酪氨酸激酶相关的活性,所述酶使磷脂酰肌醇(pi)和其在肌醇环的3′-羟基处的磷酸化衍生物磷酸化(panayotou等人(1992)trendscellbiol2:358-60)。磷酸肌醇3-激酶(pi3k)是在肌醇环的3-羟基残基处使脂质磷酸化的脂质激酶(whitman等人(1988)nature,332:664)。通过pi3-激酶生成的3-磷酸化磷脂(pip3)充当募集具有脂质结合结构域(包括普列克底物蛋白(plekstrin)同源(ph)区)的激酶的第二信使,所述激酶例如akt和pdk1(磷酸肌醇依赖性激酶-1)(vivanco等人(2002)naturerev.cancer2:489;phillips等人(1998)cancer83:41)。
pi3激酶家族包括根据结构同源性加以细分的至少15种不同的酶,并基于序列同源性和通过酶催化形成的产物分为3类。i类pi3激酶由2个亚基组成:110kd催化亚基和85kd调节亚基。调节亚基含有sh2结构域并与被具有酪氨酸激酶活性的生长因子受体磷酸化的酪氨酸残基或致癌基因产物结合,从而诱导磷酸化其脂质底物的p110催化亚基的pi3k活性。i类pi3激酶涉及细胞因子,整合素,生长因子和免疫受体下游的重要信号转导事件,这表明控制该通路可获得重要的治疗作用,如调节细胞增殖和致癌作用。i类pi3k可使磷脂酰肌醇(pi),磷脂酰肌醇-4-磷酸和磷脂酰肌醇-4,5-二磷酸(pip2)磷酸化,分别产生磷脂酰肌醇-3-磷酸(pip),磷脂酰肌醇-3,4-二磷酸和磷脂酰肌醇-3,4,5-三磷酸。ii类pi3k磷酸化pi和磷脂酰肌醇-4-磷酸。iii类pi3k只能磷酸化pi。癌症中的关键pi3激酶同等型是i类pi3激酶,即p110α,如p110α中的复发性致癌突变指示的(samuels等人(2004)science304:554;us5824492;us5846824;us6274327)。其他同等型在癌症中可能是重要的,并且也涉及心血管和免疫炎性疾病(workmanp(2004)biochemsoctrans32:393-396;patel等人(2004)proc.am.assoc.ofcancerres.(abstractlb-247)95thannualmeeting,march27-31,orlando,florida,usa;ahmadik和waterfieldmd(2004)“phosphoinositide3-kinase:functionandmechanisms”encyclopediaofbiologicalchemistry(lennarzwj,lanemdeds)elsevier/academicpress)。
继ras之后,pi3k是癌症中第二重要的突变的致癌基因。在结肠,乳腺,脑,肝,卵巢,胃,肺和头颈部实体瘤中发现显著频率的p110α的致癌突变。大约35-40%的激素受体阳性(hr+)乳腺癌肿瘤存在pik3ca突变。在胶质母细胞瘤,黑素瘤,前列腺癌,子宫内膜癌,卵巢癌,乳腺癌,肺癌,头颈部癌,肝细胞癌和甲状腺癌中也发现pten异常。磷酸酶与张力蛋白同源物(pten)是在人类中由pten基因编码的蛋白质(steckpa等人(1997)nat.genet.15(4):356-62)。该基因的突变是许多癌症发展的一个步骤。pten通过其磷酸酶蛋白产物的作用而充当肿瘤抑制基因。这种磷酸酶涉及细胞周期的调节,防止细胞生长和分裂得太快(chuec等人(2004)med.sci.monit.10(10):ra235-41)。
pi3激酶是由p85和p110亚基组成的异二聚体(otsu等人(1991)cell65:91-104;hiles等人(1992)cell70:419-29)。已经鉴定了四种不同的i类pi3k同等型,命名为pi3kα(阿尔法),β(贝塔),δ(德尔塔),和γ(伽马),每种由独特的110kda催化亚基和调节亚基组成。三个催化亚基,即p110α,p110β和p110δ,各自与相同的调节亚基p85相互作用;而p110γ与独特的调节亚基p101相互作用。这些pi3k中的每一种在人细胞和组织中的表达模式是独特的。在每个pi3kα,β和δ同等型亚型中,p85亚基通过其sh2结构域与靶蛋白中的磷酸化酪氨酸残基(存在于适当的序列上下文中)的相互作用而将pi3激酶定位于质膜上(rameh等人(1995)cell,83:821-30;volinia等人(1992)oncogene,7:789-93)。
测量生物标志物的水平(例如,血浆中分泌蛋白质的表达水平或功能蛋白水平)可以是鉴定将对特定疗法作出响应的患者和患者群体的有效手段,所述疗法包括例如用化疗剂进行治疗。需要更有效的手段来确定哪些患有过度增殖疾病如癌症的患者将对化疗剂的治疗作出响应,并且将这些确定结合到患者的更有效的治疗方案中:化疗剂是用作单一药剂还是与其他药剂联合。
磷酸肌醇3-激酶(pi3k)信号传导级联,为细胞存活,生长和代谢的关键介质,在人类癌症中频繁改变。在大约30%的乳腺癌中发生pik3ca(编码pi3k的α-催化亚基的基因)突变的激活。这些突变导致所述酶的组成性活性并且是致癌的。突变体pik3cah1047r在乳腺导管腔上皮细胞中的表达可以诱发异质性肿瘤,所述异质性肿瘤表达管腔和基底标志物并且为雌激素受体阳性。pik3cah1047r癌基因靶向多能祖细胞,并重现具有pik3cah1047r的人乳腺肿瘤的特征(meyer等人(2011)cancerres;71(13):4344-51)。pi3k的过度活化可由于pik3ca(编码pi3k的p110α亚基的基因)的体细胞突变而发生。her2癌基因在所有乳腺癌的25%中被扩增出,这些肿瘤中的一些也包含pik3ca突变。pi3k可以增强转化并导致对her2定向疗法的抗性。在也过表达her2的mcf10a人乳腺上皮细胞中引入pi3k突变e545k和h1047r对mcf10a/her2细胞赋予了功能增强。h1047rpi3k而不是e545kpi3k的表达显著上调了her3/her4配体遗传调节蛋白(hrg)(chakrabarty等人(2010)oncogene29(37):5193-5203)。
pi3激酶/akt/pten通路对于癌症药物开发是有吸引力的靶标,因为预期这些药剂可以抑制细胞增殖,遏制来自为癌细胞提供存活和化学抗性的基质细胞的信号,逆转细胞凋亡的遏制,并克服癌细胞对细胞毒性剂的固有抗性。某些噻吩并嘧啶化合物具有p110α结合,pi3激酶抑制活性,并抑制癌细胞的生长(wallin等人(2011)mol.can.ther.10(12):2426-2436;sutherlin等人(2011)jour.med.chem.54:7579-7587;us2008/0207611;us7846929;us7781433;us2008/0076758;us7888352;us2008/0269210。gdc-0941(匹克替利昔布(pictilisib),cas登记号957054-30-7,genentechinc.)是pi3k的选择性口服生物利用度抑制剂,具备有前途的药代动力学和制药学特性(folkes等人(2008)jour.ofmed.chem.51(18):5522-5532;us7781433;us8324206;belvin等人,americanassociationforcancerresearchannualmeeting2008,99th:april15,abstract4004;folkes等人,americanassociationforcancerresearchannualmeeting2008,99th:april14,abstractlb-146;friedman等人,americanassociationforcancerresearchannualmeeting2008,99th:april14,abstractlb-110;wallin等人(2012)clin.cancerres.18:3901-3911;yuan等人(2013)oncogene32:318-326;o′brien等人(2010)clincancerres.16:3670-3683;salphati等人(2010)drugmetab.anddisp.38(9):1436-1442;edgar等人(2010)cancerres.70:1164-1172)并且与某些化疗剂联合显示出抗实体瘤细胞系的体外和体内协同活性(us8247397;us8604014;us8536161)。
他塞利昔布(gdc-0032,genentechinc.,rocherg7604,cas登记号1282512-48-4),命名为2-(4-(2-(1-异丙基-3-甲基-1h-1,2,4-三唑-5-基)-5,6-二氢苯并[f]咪唑并[1,2-d][1,4]氧氮杂卓-9-基)-1h-吡唑-1-基)-2-甲基丙酰胺,是pi3kα(阿尔法)的选择性的有力的口服生物利用度抑制剂,具有ki=0.2nm,并且对pi3kβ(贝塔)具有降低的抑制活性(ndubaku等人(2013)jour.med.chem.56(11):4597-4610;staben等人(2013)bioorg.med.chem.lett.23:2606-2613;wo2011/036280;us8242104;us8343955;us8586574;us2014/0044706)。正在患有局部晚期或转移性实体瘤的患者中进行他塞利昔布的研究。相对于pik3ca突变体异种移植物中的泛i类pi3k抑制剂gdc-0941(genentechinc.,匹克替利昔布),这种选择性分布和优异的药代动力学和药学特性允许在最大耐受剂量下获得更好的体内功效。磷酸肌醇-3激酶α同等型(pik3ca)的突变常见于乳腺癌,所述突变使pi3k信号传导通路激活(kang等人(2005)cellcycle4(4):578-581)。突变使脂质结合增加,其中螺旋突变通过削弱与p85亚基的抑制性相互作用而激活(zhao和vogt(2008)oncogene27(41):5486-5496)。激酶结构域突变通过改变蛋白质构象而激活(burke等人(2012)proc.natl.acad.sci.109(38):15259-15264)。值得注意的是,相对于具有野生型pi3k的细胞,gdc-0032优先抑制pik3ca突变体细胞。gdc-0032有力抑制pi3k下游的信号转导,并在乳腺癌细胞系和包含pik3ca突变的异种移植模型中以低浓度诱导细胞凋亡。gdc-0032的突变体偏差与gdc-0032的独特性质相关,所述性质包括针对突变体同等型的细胞效力和受体酪氨酸激酶(rtk)信号传导的减少。
他塞利昔布是i类pi3kα,-δ和-γ同等型的有效的选择性抑制剂,相比于对野生型pi3kα而言,其对突变体pi3kα同等型显示出更好的选择性(oliveroa等人,americanassociationforcancerresearchannualmeeting,washington,dc,usa,april6-10,2013;wallinj等人,36thsanantoniobreastcancersymposium,sanantonio,tx,usa,december10-14,2013)。在pik3ca突变的乳腺癌(bc)模型中,他塞利昔布增强了包括er拮抗剂氟维司群在内的标准护理治疗剂的功效(sampathd等人,36thsanantoniobreastcancersymposium(sabcs),sanantonio,tx,usa,dec.10-14,2013)。pik3ca突变是乳腺癌(bc)中最常见的基因组改变之一,存在于约40%的雌激素受体(er)阳性,her2阴性乳腺肿瘤中。pik3ca突变促进肿瘤的生长和增殖,并介导bc对内分泌治疗的抗性。相比于对野生型pi3kα(alpha)而言,他塞利昔布对突变体pi3kα显示出更好的选择性。他塞利昔布对pik3ca突变的乳腺癌细胞系具有增强的活性,临床数据包括经过确认的用他塞利昔布作为单一药物或与氟维司群联合进行治疗的pik3ca突变型bc患者的部分响应。
发明概述
他塞利昔布(gdc-0032,genentechinc.,rocherg7604,cas登记号1282512-48-4),命名为2-(4-(2-(1-异丙基-3-甲基-1h-1,2,4-三唑-5-基)-5,6-二氢苯并[f]咪唑并[1,2-d][1,4]氧氮杂卓-9-基)-1h-吡唑-1-基)-2-甲基丙酰胺,其诱导突变型p110α蛋白的降解。pi3k抑制剂降解p110的能力与临床反应相关。p110α蛋白的耗竭可以鉴定出对他塞利昔布治疗将会作出响应的患者。
他塞利昔布以剂量,时间,泛素和蛋白酶体依赖性方式特异性地促进突变型p110α亚基的降解。迄今为止,对测试的所有p110突变观察到的这种效应可以通过p110-p85相互作用的失稳而介导。
突变体而不是野生型pi3k蛋白的降解似乎阻止了rtk驱动的pi3k通路的再激活,从而可能使使用pi3k抑制剂的治疗窗更宽。
本发明的一个方面是选择用他塞维昔布进行治疗的患者的方法,该方法包括:
(a)用他塞利昔布处理从患者获得的生物样品;和
(b)检测p110α蛋白的耗竭;
其中生物样品中p110α蛋白的耗竭鉴定出对他塞利昔布治疗将会作出响应的患者。p110α蛋白的耗竭指示患者对化合物的治疗响应性,并且可以通过与抗p110α抗体的结合加以量度。通过western印迹分析,酶联免疫吸附测定(elisa),放射免疫测定(ria),免疫组织化学(ihc),荧光激活细胞分选(facs)或反相蛋白质阵列来确定抗p110α抗体与样品中p110α蛋白的结合。
本发明的一个方面是治疗患者的方法,该方法包括:
(a)测试针对pik3ca突变状态从患者获得的生物样品,其中所述pik3ca突变状态包括选自h1047r,c420r,h1047l,e542k,e545k和q546r的突变;
(b)使来自具有pik3ca突变的患者的生物样品与他塞利昔布接触并检测p110α同等型的耗竭;和
(c)向具有pik3ca突变的患者施用他塞利昔布。所述生物样品可以是循环肿瘤细胞。与他塞利昔布联合,可以给患者施用选自5-fu,多西他赛,艾日布林,吉西他滨,gdc-0973,gdc-0623,帕利他赛,他莫昔芬,氟维司群,地塞米松,帕妥珠单抗,曲妥珠单抗-艾美坦辛,曲妥珠单抗和来曲唑的化疗剂。患者可能患有表达her2的乳腺癌或雌激素受体阳性(er+)乳腺癌。癌症可能是转移性的。可以在辅助治疗中向患者施用他塞利昔布。
本发明的一个方面是选择具有pik3ca突变的以便用他塞维昔布进行治疗的患者的方法,该方法包括:
(a)检测从患者获得的生物样品中的pik3ca突变;和
(b)将施用他塞利昔布之前从患者获得的生物样品中的p110α水平与施用他塞利昔布之后从所述患者获得的生物样品中的p110α水平进行比较,
其中在施用他塞利昔布之后从患者获得的生物样品中p110α的水平的耗竭鉴定出对他塞利昔布治疗将会作出响应的患者。
本发明的一个方面是治疗癌症的方法,该方法包括:
(a)将施用他塞利昔布之前从癌症患者获得的生物样品中的p110α水平与施用他塞利昔布之后从所述患者获得的生物样品中的p110α水平进行比较,和
(b)改变施用于患者的他塞利昔布治疗的剂量,给药频率,或疗程。
本发明的一个方面是监测癌症患者中的治疗功效的方法,该方法包括:(a)向所述患者施用他塞利昔布;
(b)测定在施用他塞利昔布之后从所述患者获得的生物样品中的p110α;和
c)改变施用于患者的他塞利昔布治疗的剂量,给药频率,或疗程。
本发明的一个方面是为诊断患有癌症的患者选择治疗方案的方法,所述方法包括使所述患者的癌细胞与有效量的他塞利昔布接触,并检测响应于他塞利昔布的p110α水平,其中p110α耗竭的检出指示所述癌症对他塞利昔布治疗易感,并且,其中所述治疗方案包括:如果确定所述癌症对他塞利昔布治疗易感,则向所述患者施用他塞利昔布。
癌细胞可能是pik3ca突变型癌细胞。
本发明的一个方面是治疗癌症的方法,该方法包括:
a)向患者施用他塞利昔布;
b)测定从所述患者获得的生物样品中的p110α水平或与p110α水平相关的生物标志物的水平的变化;和
c)选择有待施用于患者的他塞利昔布治疗的剂量,给药频率,或疗程,所述患者显示在从所述患者获得的生物样品中p110α的耗竭。p110α水平的变化可以是p110α水平的耗竭。
本发明的一个方面是鉴定用于监测在癌症治疗中对他塞利昔布的响应性的生物标志物的方法,该方法包括:
(a)检测与从接受了至少一个剂量的他塞利昔布的患者获得的生物样品中的p110α水平相关的生物标志物的表达,调控或活性;和
(b)将所述生物标志物的表达,调控或活性与参考样品中的生物标志物状态进行比较,其中所述参考样品是在施用他塞利昔布之前从患者获得的生物样品;
其中其调控与参考样品相比的改变为低或高至少2倍的生物标志物被鉴定为用于监测对他塞利昔布的响应性的生物标志物。癌症可能是表达her2的乳腺癌。
本发明的一个方面是治疗患者中的癌症的方法,该方法包括向患者施用治疗有效量的他塞利昔布,其中治疗的基础是与从所述患者获得的生物样品中p110α水平相关的生物标志物的检测。生物样品可以是肿瘤活检样品或循环肿瘤细胞。
附图简述
图1显示通过他塞利昔布降解的p110α蛋白的western印迹分析对于pi3kα突变型细胞而言是剂量依赖性的,特异的。这种作用机制允许他塞利昔布通过rtk而减少反馈的影响,否则会减弱抗肿瘤活性。
图2显示了用他塞利昔布,匹克替利昔布(gdc-0941,genentech)和阿哌利昔布(alpelisib)(byl719,novartiscas#:1217486-61-7)处理hcc1954细胞(pik3cah1047r)24小时的western印迹分析。其他口服pi3k抑制剂,匹克替利昔布和阿哌利昔布在临床开发中未降解突变型p110α蛋白。
图3显示了用他塞利昔布处理的pik3ca野生型hdqp1乳腺癌细胞和突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。他塞利昔布导致pik3ca突变型细胞系中p110α耗竭,而不影响p85水平。
图4显示了使用各种浓度(0.2μm,1μm,5μm)的他塞利昔布以及对照(dmso溶媒)的sw48等基因系的western印迹分析,所述sw48等基因系包括sw48亲本pik3ca野生型,突变型等基因sw48e545k杂合子(het)和pik3ca突变型等基因sw48h1047r杂合子细胞。
图5a显示了细胞中p110αmrna表达的绘图,其通过相对于18srna水平的相对p110αmrna表达加以量度。药物未改变p110α的mrna表达。
图5b显示了产生sw48e545k半合子系的crispr(成簇的规律间隔的短回文重复序列)的western印迹分析(两个克隆重复运行)。该western印迹显示,与sw48e545k杂合系相比,突变型p110α水平显著降低,这表明突变型p110α可能比wtp110α更不稳定。从左到右的泳道是sw48e545k半合子克隆1,sw48e545k半合子克隆2,sw48亲本,sw48e545k杂合子,sw48e545k杂合子。
图6显示了用他塞利昔布1μm和5μm以及对照(dmso溶媒)处理的pik3ca突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。p110alpha(p110α)以时间依赖性方式耗竭。
图7a显示了在用他塞利昔布(gdc0032)处理的hcc1954野生型(左)和h1047r突变型(右)p110α细胞中相对于18s对照测量的相对rna水平的实时qpcr结果。
图7b显示了在用他塞利昔布(gdc0032)处理的hcc1954p110α野生型(左)和p110αh1047r突变体(右)p110α细胞中相对于rpl19对照测量的p110αmrna表达的实时qpcr结果。
图7c显示了在用阿哌利昔布(byl-719)处理的hcc1954p110α野生型(左)和p110αh1047r突变体(右)p110α细胞中相对于rpl19对照测量的p110αmrna表达的实时qpcr结果。
图8a显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132(右侧的泳道)。
图8b显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞裂解物的western印迹分析。在收获之前4小时,加入10μmmg132(中间的泳道)和10μmuae1抑制剂。
图8c显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132(右侧的泳道)。
图8d显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132(右侧的泳道)。
图8e显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132,氯喹或氯化铵(右侧的泳道)。
图8f显示了p110泛素化的途径图,描述了动物细胞通过路径特异性抑制剂降解蛋白质的两种不同机制。
图9a显示了用他塞利昔布和阿哌利昔布(byl719)处理的pi3k野生型braf突变型sw982细胞的western印迹分析。他塞利昔布未使p110δ降解或耗竭。
图9b显示了用g-102(表1,us8242104)和gdc-0032(他塞利昔布)处理的pi3k野生型b细胞淋巴瘤su-dhl-10细胞的western印迹分析。
图10显示了以不同浓度(100nm,1μm,5μm)的pi3k抑制剂以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的pik3ca野生型hdqp1乳腺癌细胞的western印迹分析。
图11a显示了以不同浓度(3nm,16nm,60nm,400nm,2μm)的g-102(表1)以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的mda-mb453(h1047r)细胞的western印迹分析。
图11b显示了以不同浓度(3nm,16nm,60nm,400nm,2μm)的他塞利昔布(gdc-0032)以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的mda-mb453(h1047r)细胞的western印迹分析。
图12显示了以不同浓度(1nm,10nm,100nm)的他塞利昔布(gdc-0032)和g-102(表1)以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的sw48h1047r细胞的western印迹分析。
图13显示了以不同浓度(1nm,10nm,100nm)的他塞利昔布(gdc-0032)和g-102(us8242104)以及对照(dmso溶媒)进行pi3k抑制处理的具有和没有生长因子配体nrg的sw48pik3cah1047r细胞的western印迹分析。
图14a显示了以不同浓度的g-181处理24小时时相对于野生型亲本pi3k细胞的等基因突变型(e545k,h1047r)细胞中ppras40的体外细胞增殖,以建立亲本/突变体选择性ec50值。
图14b显示了以不同浓度的gdc-0032处理24小时时相对于野生型亲本pi3k细胞的等基因突变型(e545k,h1047r)细胞中ppras40的体外细胞增殖,以建立亲本/突变体选择性ec50值。
图14c显示了以不同浓度的g-102处理24小时时相对于野生型亲本pi3k细胞的等基因突变型(e545k,h1047r)细胞中ppras40的体外细胞增殖,以建立亲本/突变体选择性ec50值。
图15显示了sw48等基因野生型和突变型(e545k,h1047r)细胞系的体外细胞增殖数据以及用剂量逐步增高的他塞利昔布和泛pi3k抑制剂匹克替利昔布(gdc-0941)进行处理的绘图。
图16显示了匹克替利昔布(gdc-0941),bkm120(布帕利昔布(buparlisib),novartisag,cas登记号944396-07-0),他塞利昔布(gdc-0032)和byl719(阿哌利昔布,novartiscas号1217486-61-7)在针对pik3ca突变型细胞系的4天细胞增殖(cell-titerglor,promega)测定中的功效(ic50微摩尔)的绘图。每个圆点代表不同的癌细胞系。
图17显示了利用细胞死亡-核小体elisa检测,在72小时研究中pik3ca野生型和突变型(e545k,h1047r)细胞系的体外细胞增殖数据以及用剂量逐步增高的他塞利昔布和pi3kα选择性抑制剂gdc-0326(us8242104)和byl719进行处理的绘图。
图18a显示了荷载包含pik3cah1047r(pi3kα)突变的hcc1954.x1乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过21天的拟合的肿瘤体积变化,所述小鼠每天通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80),150mg/kg的匹克替利昔布(gdc-0941)和25mg/kg的他塞利昔布(gdc-0032)。术语μl表示微升。
图18b显示了荷载包含pik3cah1047r(pi3kα)突变的hcc1954.x1乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过21天的拟合的肿瘤体积变化,所述小鼠每天通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80),40mg/kg的阿哌利昔布(byl-719)和15mg/kg的他塞利昔布(gdc-0032)。
图18c显示了荷载包含pik3cae542k(pi3kα)突变的源于whim20激素受体阳性患者的乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过28天的拟合的肿瘤体积变化,所述小鼠每天通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80)和15mg/kg的他塞利昔布(gdc-0032)。
图18d显示了荷载包含pik3cah1047r(pi3kα)突变的源于hci-003激素受体阳性患者的乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过27天的拟合的肿瘤体积变化,所述小鼠每天通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80),40mg/kg的阿哌利昔布(byl-719)以及2.5,5.0,15mg/kg的他塞利昔布(gdc-0032)。
图19显示了用不同浓度(16nm,80nm,400nm)他塞利昔布以及对照(dmso溶媒)处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。
图20显示了h0147r突变型p110αpi3k同等型的激酶结构域的构象的模型。
图21显示了在包含pik3ca突变体的细胞系组的四天活力测定中的gdc-0032效力(ic50)的绘图。根据pik3ca突变的位置显示数据。
图22显示了p85与p110α的免疫共沉淀(co-ip)的western印迹(wb)分析,表明稳定的p110α与p85复合的与p110α平行的水平以及由他塞利昔布诱导的显著剂量依赖性p110α降解。
图23显示了稳态p110αmrna表达。
图24a显示了根据实施例7的野生型pik3cahcc-1954(上)和表达pik3cahcc-1954的h1047r突变(下)的胰蛋白酶切割。
图24b显示了根据实施例7的野生型pik3cahcc-1954(左)和表达pik3cahcc-1954的h1047r突变(右)在消化和p110α(pik3ca)蛋白免疫沉淀之后的液相色谱-串联质谱(lc-ms/ms)分析。
示例性实施方案的详述
现在将详细参考本发明的某些实施方案,其实施例展示在所附的结构和公式中。虽然将结合列举的实施方案描述本发明,但是应当理解,它们并不旨在将本发明限于那些实施方案。相反,本发明旨在覆盖可以包括在权利要求书所限定的本发明范围内的所有替代方案,修改和等同物。本领域技术人员将认识到可以用于本发明实践中的与本文所述那些类似或等同的许多方法和材料。本发明决不限于所描述的方法和材料。在并入的文献,专利和相似材料中的一者或多者与本申请不相同或相矛盾的情况下,包括但不限于定义的术语,术语使用,所描述的技术等等在内,以本申请为准。
定义
在本说明书和权利要求中使用时,词语“包含”("comprise"),“包含”("comprising"),“包括”("include"),“包括”("including")和“包括”("includes")旨在规定所述特征,整体,部件或步骤的存在,但是它们不排除存在或添加一个或多个其他特征,整体,部件,步骤或其组。
术语“治疗”("treat")和“治疗”("treatment")是指治疗性治疗和预防性或防止性措施,其目的是预防或减缓(减轻)不期望的生理变化或失调,例如癌症的生长,发展或扩散。出于本发明的目的,有益或期望的临床结果包括但不限于:症状的减轻,疾病程度的降低,疾病状态的稳定化(即,未恶化),疾病进展的延迟或减缓,疾病状态的改善或减轻,以及缓解(不管是部分还是全部),无论是可检测的还是不可检测的。如果没有接受治疗,“治疗”也可能意味着相比于预期生存的生存期延长。需要治疗者包括已经患有病症或失调者以及具有患该病症或失调的倾向者,或其中有待预防该病症或失调者。
短语“治疗有效量”是指(i)治疗特定疾病,病症或失调,(ii)减弱,改善或消除特定疾病,病症或失调的一种或多种症状,或(iii)预防或延迟本文所述的特定疾病,病症或失调的一种或多种症状发作的本发明化合物的量。在癌症的情况下,药物的治疗有效量可以减少癌细胞的数量;减小肿瘤大小;抑制(即,在一定程度上减慢并优选地停止)癌细胞浸润到周围器官中;抑制(即,在一定程度上减慢并优选停止)肿瘤转移;在一定程度上抑制肿瘤生长;和/或在一定程度上减轻与癌症相关的一种或多种症状。在药物可以预防现有癌细胞生长和/或将其杀死的情况下,所述药物可能是细胞生长抑制的和/或细胞毒性的。对于癌症治疗,可以例如通过评定疾病进展时间(ttp)和/或确定缓解率(rr)来量度功效。
术语“生物样品”包括取自动物或人的组织,细胞和体液的所有样品。
患者的“有效反应”或患者对药物治疗的“响应性”和类似措辞是指对患者赋予的临床益处或治疗益处,所述患者具有疾病或失调(如癌症)的风险,或患有所述疾病或失调。在一个实施方案中,这种益处包括以下任何一者或多者:延长生存(包括总生存期和无进展生存期);获得客观响应(包括完全响应或部分响应);或改善癌症的体征或症状。在一个实施方案中,使用生物标志物(例如,使用ihc确定的突变型p110α)来鉴定相对于不表达该生物标志物的患者预期具有对药物例如他塞利昔布(gdc-0032)治疗有响应的可能性增加的患者。在一个实施方案中,使用生物标志物(如使用ihc确定的)来鉴定相对于不表达相同水平的该生物标志物的患者预期具有对药物治疗有响应的可能性增加的患者。在一个实施方案中,使用生物标志物的存在来鉴定相对于不存在该生物标志物的患者更可能对药物治疗作出响应的患者。在另一个实施方案中,使用生物标志物的存在来确定相对于不存在该生物标志物的患者将具有从药物治疗中受益可能性增加的患者。
与癌症(例如乳腺癌或nsclc)患者临床益处增加相关联的生物标志物的“量”或“水平”是指生物样品中的可检测水平,其中生物标志物的水平与患者临床益处增加相关联。这些可以通过本领域专业技术人员已知的并且也被本发明公开的方法进行测量。评定的生物标志物的表达水平或量可用于确定对治疗的响应。在一些实施方案中,利用(例如,来自活检或血液的患者肿瘤样品的)ihc来确定生物标志物的量或水平。在一些实施方案中,与癌症患者的临床益处增加相关联的生物标志物的量或水平是ihc评分2,ihc评分3,或ihc评分2或3。在一些实施方案中,与癌症患者的临床益处增加相关联的c-met生物标志物的量或水平为:50%或更多肿瘤细胞具有中等染色强度,组合的中等/高染色强度,或高染色强度。在一些实施方案中,与癌症患者的临床益处增加相关联的生物标志物的量或水平为:50%或更多肿瘤细胞具有中等染色强度或高染色强度。
术语“检测”包括任何检测手段,包括直接检测和间接检测。
术语“诊断”在本文中用于指分子状态或病理状态,疾病或病症的鉴定或分类。例如,“诊断”可以指特定类型的癌症例如肺癌的鉴定。“诊断”还可以指例如过组织学(例如,非小细胞肺癌),通过分子特征(例如,通过在特定基因或蛋白质中的核苷酸和/或氨基酸变异表征的肺癌),或通过这两者进行的具体癌症类型的分类。
本文中使用的术语“预后”是指可归因于癌症的死亡或进展的可能性的预测,包括例如肿瘤疾病如癌症的复发,转移扩散和抗药性。
术语“预测”("prediction")(以及诸如变型“预测”(predicting))在本文中用于指患者有利或不利地对药物或药物集合作出响应的可能性。在一个实施方案中,预测涉及这些反应的程度。在另一个实施方案中,预测涉及患者在治疗之后是否将存活和/或存活的概率,所述治疗例如在没有癌症复发的情况下用特定治疗剂治疗和/或手术切除原发性肿瘤和/或化疗一段时间。本发明的预测方法可在临床上用于通过为任何特定患者选择最适当的治疗方式做出治疗决定。本发明的预测方法是预测患者是否可能有利地对治疗方案(例如给定的治疗方案)做出反应或者预测患者在治疗方案之后是否可能长期生存的有价值的工具,所述治疗方案包括例如施用给定治疗剂或组合,外科手术,化学治疗等。
当根据本发明使用时,针对特定治疗剂或治疗选项的术语“抗性增加”是指对标准剂量的药物或对标准治疗方案的响应降低。
当根据本发明使用时,针对特定治疗剂或治疗选项的术语“敏感性降低”是指对标准剂量的药剂或标准治疗方案的响应降低,其中响应降低可以通过增加药剂的剂量或治疗的强度加以补偿(至少部分地补偿)。
可以使用任何指示患者益处的终点来评估“患者响应”,所述终点包括但不限于(1)在一定程度上抑制肿瘤生长,包括减缓或完全生长停滞;(2)减少肿瘤细胞数;(3)减小肿瘤大小;(4)抑制(例如,减少,减缓或完全停止)肿瘤细胞浸润到相邻周围器官和/或组织中;(5)抑制(例如减少,减缓或完全停止)转移;(6)增强抗肿瘤免疫反应,其可能但不一定导致肿瘤消退或排斥;(7)在一定程度上减轻与肿瘤相关的一种或多种症状;(8)延长治疗后生存期;和/或(9)在治疗后的给定时间点的死亡率降低。
“生物标志物”是客观测量和评估作为针对治疗干预的正常生物过程,致病过程或药理学反应的指标的特征。生物标志物可有几种类型:预测性的,预后性的或药效学(pd)生物标志物。预测性生物标志物预测哪些患者可能会对特定治疗作出响应或从中获益。预后性生物标志物预测患者疾病的可能过程,并可以指导治疗。药效学生物标志物证实药物活性,并能够优化剂量和施用日程表。
通过使用普遍用于建立药效学(pd)的一种或多种方法分析生物样品,检测在体外或体内发生的生物标志物状态的“变化”或“调控”,包括pik3ca突变或pik3ca突变的集合,所述方法包括:(1)对生物样品的基因组dna或逆转录pcr产物进行测序,由此检测到一个或多个突变;(2)通过定量信息水平或估计拷贝数来评估基因表达水平;和(3)通过免疫组织化学,免疫细胞化学,elisa或质谱术分析蛋白质,由此检测蛋白质的降解,稳定化或翻译后修饰,例如磷酸化或泛素化。
术语“癌症”和“癌性的”是指或描述在哺乳动物中典型地以不受调节的细细胞生长为特征的生理状况。“肿瘤”包含一个或多个癌细胞。癌症的实例包括但不限于,癌,淋巴瘤,母细胞瘤,肉瘤,以及白血病或淋巴系统恶性肿瘤。这些癌症的更具体的实例包括鳞状细胞癌(例如上皮鳞状细胞癌);肺癌,包括小细胞肺癌,非小细胞肺癌(“nsclc”),肺腺癌和肺鳞状细胞癌;腹膜癌;肝细胞癌;胃的癌或胃癌,包括胃肠癌;胰腺癌;胶质母细胞瘤;宫颈癌;卵巢癌;肝的癌;膀胱癌;肝细胞瘤;乳腺癌;结肠癌;直肠癌;结肠直肠癌;子宫内膜癌或子宫癌;唾液腺癌,肾的癌或肾癌;前列腺癌;外阴癌;甲状腺癌;肝癌;肛门癌;阴茎癌;以及头颈癌。如本文中使用的胃部癌包括胃癌,其可以在胃的任何部分发展,并且可以扩散至整个胃并且到达其他器官,特别是食道,肺,淋巴结和肝脏。
术语“造血系统恶性肿瘤”是指在涉及细胞如白细胞,淋巴细胞,自然杀伤细胞,浆细胞和骨髓细胞如嗜中性粒细胞和单核细胞的造血过程中产生的癌症或过度增殖性疾病。造血系统恶性肿瘤包括非霍奇金淋巴瘤,弥漫性大造血淋巴瘤,滤泡性淋巴瘤,套细胞淋巴瘤,慢性淋巴细胞白血病,多发性骨髓瘤,急性髓性白血病和髓细胞白血病。淋巴细胞白血病(或“淋巴母细胞白血病”)包括急性淋巴母细胞白血病(all)和慢性淋巴细胞白血病(cll)。髓性白血病(也称“髓样”或“非淋巴细胞性”)包括急性髓性(或成髓细胞性)白血病(aml)和慢性髓性白血病(cml)。
“化疗剂”是用于治疗癌症的生物(大分子)或化学(小分子)化合物,无论其作用机理如何。
术语“哺乳动物”包括但不限于,人,小鼠,大鼠,豚鼠,猴,狗,猫,马,牛,猪和绵羊。
术语“包装说明书”用于指通常包括在治疗产品的商业包装中的说明书,其包含关于使用这种治疗产品的适应症,用法,剂量,给药,禁忌症和/或警告的信息。
如本文中使用的短语“药学上可接受的盐”是指本发明化合物的药学上可接受的有机盐或无机盐。示例性的盐包括但不限于,硫酸盐,柠檬酸盐,乙酸盐,草酸盐,氯化物,溴化物,碘化物,硝酸盐,硫酸氢盐,磷酸盐,酸式磷酸盐,异烟酸盐,乳酸盐,水杨酸盐,酸式柠檬酸盐,酒石酸盐,油酸盐,鞣酸盐,泛酸盐,酒石酸氢盐,抗坏血酸盐,琥珀酸盐,马来酸盐,龙胆酸盐,富马酸盐,葡糖酸盐,葡萄糖醛酸盐,蔗糖盐,甲酸盐,苯甲酸盐,谷氨酸盐,甲磺酸酯“甲磺酸盐”,乙磺酸盐,苯磺酸盐,对甲苯磺酸盐和双羟萘酸盐(即1,1′-亚甲基双-(2-羟基-3-萘甲酸))盐。药学上可接受的盐可涉及包括另外的分子,例如乙酸根离子,琥珀酸根离子或其他反离子。反离子可以是使母体化合物上的电荷稳定的任何有机模块或无机模块。此外,药学上可接受的盐可以在其结构中具有多于一个的带电原子。多个带电原子是药学上可接受的盐的一部分的实例可以具有多个反离子。因此,药学上可接受的盐可以具有一个或多个带电原子和/或一个或多个反离子。
可以通过本领域可获得的任何适合的方法制备期望的药学上可接受的盐。例如,用无机酸(如盐酸,氢溴酸,硫酸,硝酸,甲磺酸,磷酸等等)或用有机酸(如乙酸,马来酸,琥珀酸,扁桃酸,富马酸,丙二酸,丙酮酸,草酸,乙醇酸,水杨酸,吡喃糖基酸如葡糖醛酸或半乳糖醛酸,α羟基酸如柠檬酸或酒石酸,氨基酸如天冬氨酸或谷氨酸,芳香酸如苯甲酸或肉桂酸,磺酸如对甲苯磺酸或乙磺酸,等等)处理游离碱。通常被认为适合于与碱性药物化合物形成药学上有用或可接受的盐的酸在例如p.stahl等人camilleg.(编辑)handbookofpharmaceuticalsalts.properties,selectionanduse.(2002)zurich:wiley-vch;s.berge等人,journalofpharmaceuticalsciences(1977)66(1)119;p.gould,internationalj.ofpharmaceutics(1986)33201217;anderson等人,thepracticeofmedicinalchemistry(1996),academicpress,newyork;remington’spharmaceuticalsciences,18thed.,(1995)mackpublishingco.,eastonpa;以及在theorangebook(food&drugadministration,washington,d.c,在其网站上)中进行了论述。这些公开内容通过对其提述并入本文。
短语“药学上可接受的”表示该物质或组合物必须与构成制剂的其他成分和/或用其进行治疗的哺乳动物在化学上和/或毒理学上相容。
如本文中使用的术语“协同”是指比两种或更多种单一药剂的叠加效应更有效的治疗联合。基于从本文所述的测定法获得的结果,可以确定在突变体选择性,结合pi3k的化合物或其药学上可接受的盐与一种或多种化疗剂之间的协同相互作用。可以使用chou和talalay联合方法和利用calcusyn软件的剂量-效应分析来分析这些测定法的结果,以获得联合指数(chou和talalay,1984,adv.enzymeregul.22:27-55)。联合治疗可以提供“协同作用”并证明“协同”,即当活性成分一起使用时所实现的效应大于分开使用这些化合物所产生的效应的总和。可以获得协同效应,如果将活性成分:(1)共同配制为联合单位剂量制剂和同时给药或递送;(2)作为分开的制剂交替或并行递送;或(3)通过某些其他方案递送。在交替疗法中加以递送时,当将化合物依次给药或递送时,例如通过分开的注射器中的不同注射剂或分开的丸剂或片剂,可以获得协同效应。一般而言,在交替疗法期间,有效剂量的每种活性成分是依次地即连续地施用的,而在联合治疗中,有效剂量的两种或更多种活性成分是一起施用的。也可以使用bliss独立模型和最高单药(hsa)模型来评估联合效应(lehár等人,2007,molecularsystemsbiology3:80)。
“elisa”(酶联免疫吸附测定)是“湿实验室”型分析生物化学测定法的流行格式,其使用异相固相酶免疫测定(eia)的一种亚型来检测液体样品或湿样品中物质的存在(engvalle,perlmanp(1971)."enzyme-linkedimmunosorbentassay(elisa).quantitativeassayofimmunoglobuling".immunochemistry8(9):871–4;vanweemenbk,schuursah(1971)."immunoassayusingantigen-enzymeconjugates".febsletters15(3):232–236)。elisa可以实施其他形式的配体结合测定法而不是严格的“免疫”测定法,尽管由于该方法的普遍使用和发展历史,该名称保持了最初的“免疫”。该技术实质上需要任何可以固定在固相上的连接试剂以及将特异性结合酶并利用酶产生可被正确定量的信号的检测试剂。在洗涤之间,只有配体及其特异性结合对应物通过与固相的抗原-抗体相互作用而保持特异性结合或“免疫吸附”,而非特异性或未结合的组分被洗掉。不同于其他可以在洗涤之后再使用相同反应孔(例如,比色皿)的分光光度法湿法实验室测定法格式,elisa板使反应产物免疫吸附在固相上作为板的一部分,因此不便于再使用。进行elisa涉及至少一种对特定抗原具有特异性的抗体。将具有未知量抗原的样品非特异性地(通过吸附到表面上)或特异性地(在“夹心”elisa中,通过针对相同抗原特异性的另一种抗体捕获)固定在固体支持物(通常为聚苯乙烯微量滴定板)上。抗原固定后,加入检测抗体,与抗原形成复合物。检测抗体可以与酶共价连接,或者本身可以通过经由生物缀合与酶连接的二级抗体而被检测到。在每个步骤之间,通常用温和的洗涤剂溶液洗涤板以除去任何未特异性结合的蛋白质或抗体。在最后的洗涤步骤之后,通过加入酶底物产生可见信号使板显色,所述信号指示样品中抗原的量。
“免疫组织化学”(ihc)是指通过利用与生物组织中的抗原特异性结合的抗体的原理来检测组织切片的细胞中的抗原(例如,蛋白质)的过程。免疫组织化学染色广泛用于诊断异常细胞,比如在癌性肿瘤中发现的细胞。特异性分子标志物是特定细胞事件如增殖或细胞死亡(细胞凋亡)的特征。ihc也广泛用于了解生物标志物和差异表达的蛋白质在生物组织不同部分中的分布和定位。可以以多种方式实现抗体-抗原相互作用的可视化。在最常见的情况下,将抗体与能够催化生色反应的酶(例如过氧化物酶)缀合(参见免疫过氧化物酶染色)。或者,也可以将抗体标记到诸如荧光素或罗丹明的荧光团上(参见免疫荧光)。
“免疫细胞化学”(icc)是一种常见的实验室技术,其使用通过特异性表位靶向细胞中的特异性肽或蛋白质抗原的抗体。然后,可以使用几种不同的方法检测这些结合的抗体。icc可以评估特定样品中的细胞是否表达所讨论抗原。在发现免疫阳性信号的情况下,icc还确定哪些亚细胞区室正在表达抗原。
“等基因”细胞系和人类疾病模型是经选择或设计用于在体外(“在实验室中”,在人工环境中)精确模拟特定患者群体的遗传学的细胞家族。它们具备遗传匹配的“正常细胞”,以提供用于研究疾病生物学和新型治疗剂的等基因系统(bardellia等人(2003)science300(5621):949)。等基因细胞系可以用于模拟任何有关遗传基础的疾病。癌症就是这样一种疾病,已经针对其广泛使用等基因人类疾病模型。通过被称为同源基因靶向的过程产生等基因细胞系。利用同源重组的靶向载体是用于敲入或敲除有待研究的期望的致病性突变或snp(单核苷酸多态性)的工具或技术。虽然可以直接从癌症患者获得疾病突变,但是除了感兴趣的特定突变之外,这些细胞通常还包含许多背景突变,并且通常未获得匹配的正常细胞系。随后,在表征的人类癌细胞系中,使用靶向载体“敲入”或“敲除”基因突变,使得能够在两个方向上进行转换:从正常到癌基因型,或反之亦然。
术语“辅助”和“辅助治疗”是指除了初步,主要或初始治疗之外施用的护理或治疗。相对于用于癌症治疗的手术和复杂的治疗方案,该术语主要用于描述辅助癌症治疗。辅助治疗的一个实例是通常在手术后所有可检测的疾病已经消除但仍然存在由于隐匿性疾病引起的复发的统计学风险情况下施用的附加治疗。如果在手术后遗留已知的疾病,那么进一步的治疗在技术上不是辅助性的。例如,通常施用放射治疗或全身治疗作为乳腺癌手术后的辅助治疗。全身治疗包括化学疗法,免疫疗法或生物反应调节剂或激素疗法。在决定具体的辅助治疗之前,肿瘤学家使用统计学证据来评定疾病复发的风险。辅助治疗的目的是改善疾病特异性症状和总生存率。因为所述治疗实质上是针对一种风险,而不是针对可证明的疾病,所以公认的是,接受辅助治疗的一部分患者已经通过其初次手术治愈。在许多类型的癌症手术后,常常施用辅助全身治疗和放射治疗。
术语“野生型pi3kp110α同等型”是指在pi3kp110α基因中不存在突变。
术语“突变型pi3kp110α同等型”是指一个或多个激活突变位于pi3kp110α的等位基因内。
参数“ic50”是指半数最大抑制浓度,是物质在抑制特定生物功能或生物化学功能中的有效性的量度。这种定量量度指示需要多少特定的药物或其他物质(抑制剂)来抑制给定的生物过程(或过程的组分,即酶,细胞,细胞受体或微生物)的一半。它通常在药理研究中用作拮抗剂药物效力的量度。ic50代表50%体外抑制所需的药物浓度,与激动剂药物的ec50相当。ec50还表示获得体内50%最大效应所需的血浆浓度。参数ki与ic50相关(cerrz等人(2009)nucl.acidsres.37:w441-w445)。而ki是抑制剂的结合亲和力,ic50是抑制剂的功能强度。
通过他塞利昔布的p110α耗竭
他塞利昔布以剂量依赖性,时间依赖性,蛋白酶体依赖性和泛素依赖性方式使p110alpha(p110α)发生降解。在pik3ca突变型细胞中,通过他塞利昔布的降解是p110α特异性的;在野生型细胞中未观察到p85,110δ同等型或p110α的降解。通过在1小时和24小时的pakt和ppras40水平,测量到即使在反馈情况下的通路抑制的惊人和意想不到的益处。这些相关性观察结果可以扩大他塞利昔布治疗选项的治疗窗(治疗指数增加)。
在渐增剂量的他塞利昔布的存在下,他塞利昔布对p110α的降解可以通过p85的免疫沉淀(ip)/p110的western印迹进行测试,反之亦然。由于在治疗2小时的p110降解可以忽略,这是一个其中评估p110/p85解离的合理窗口,而没有使解释复杂化的显著p110降解,因而提供预测癌症患者将对他塞利昔布治疗作出响应的诊断机会。
可以通过蛋白质组学技术进行突变型与野生型p110α降解的定量,包括确定野生型和突变蛋白的半衰期,他塞利昔布在突变型和野生型细胞系中的解离率,以及泛素化位点和细胞机器的鉴定。因此,p110α的降解可以被量度为p110α的耗竭。
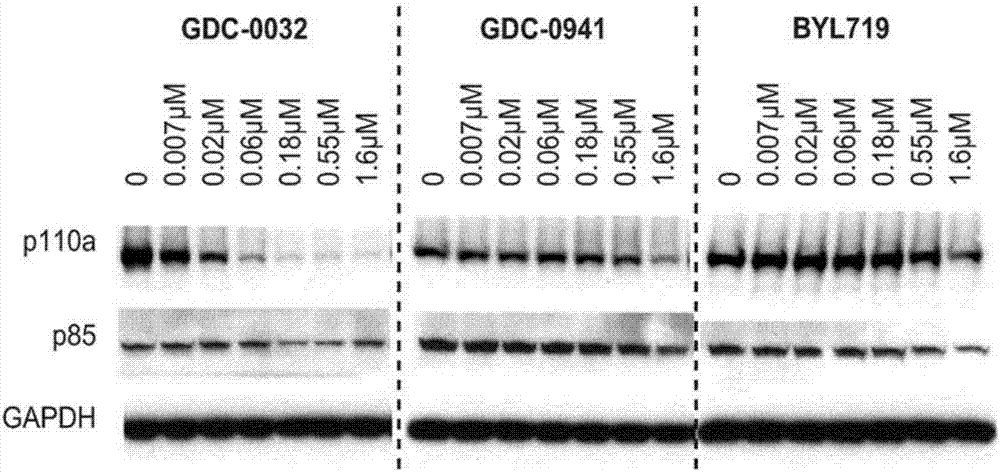
在突变型细胞中,他塞利昔布比其他pi3k抑制剂更有效,因为它唯一地使突变型p110α蛋白降解。图1显示通过他塞利昔布降解的p110α蛋白的western印迹分析对于pi3kα突变型细胞而言是剂量依赖性的,特异的。虽然本发明不受任何特定作用机制的限制,但是这种活性的原理允许他塞利昔布减少rtk反馈的影响,否则会减弱抗肿瘤活性。图2显示了用他塞利昔布,匹克替利昔布(gdc-0941,genentech)和阿哌利昔布(byl719,novartiscas#:1217486-61-7)处理hcc1954细胞(pik3cah1047r)24小时的western印迹分析。其他口服pi3k抑制剂,匹克替利昔布和阿哌利昔布在临床开发中未降解突变型p110α蛋白。
他塞利昔布导致在pik3ca突变型细胞系中的p110α耗竭,而不影响p85水平,这与导致p110α单体降解的p110α/p85的解离机制一致(图3)。当p110α作为单体从p85解离时,p110α是不稳定的,并快速转换(yu等人(1998)mol.cellbio.18:1379-1387;wu等人(2009)proc.natl.acad.sci.106(48):20258-20263)。p110α的半衰期约为1小时,而p110α/p85二聚体显著更稳定,半衰期约为5小时。
突变型p110α比野生型更易被他塞利昔布降解。图4显示了使用各种浓度(0.2μm,1μm,5μm)的他塞利昔布以及对照(dmso溶媒)的sw48等基因系的western印迹分析,所述sw48等基因系包括sw48亲本pik3ca野生型,突变型等基因sw48e545k杂合子(het)和pik3ca突变型等基因sw48h1047r杂合子细胞。
p110αe545k突变蛋白显得不如野生型蛋白稳定(图5a和5b)。在e545k工程化细胞中,突变型p110α的rna表达无变化。图5a显示了细胞中p110αmrna表达的绘图,其通过相对于18srna水平的相对p110αmrna表达加以量度。药物未改变p110α的mrna表达。图5b显示了产生sw48e545k半合子系的crispr(成簇的规律间隔的短回文重复序列)的western印迹分析(两个克隆重复运行)。该western印迹显示,与sw48e545k杂合系相比,突变型p110α水平显著降低,这表明突变型p110α可能比wtp110α更不稳定。从左到右的泳道是sw48e545k半合子克隆1,sw48e545k半合子克隆2,sw48亲本,sw48e545k杂合体,sw48e545k杂合体。
p110α以时间依赖性方式耗竭。图6显示了用他塞利昔布1μm和5μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。由于他塞利昔布具有约40小时的长临床药代动力学半衰期,正如根据患者样品所测量的,突变型p110α的降解应该正在肿瘤中发生。
虽然蛋白质减少,但他塞利昔布并不减少p110αrna。图7a显示了在hcc1954野生型和突变型p110α细胞中相对于18s对照测量的相对rna水平的实时qpcr结果。对于dmso相比于gdc-0032处理的细胞,检测到p110αmrna水平没有差异。突变体等位基因的表达升高约8倍。pik3ca的dna拷贝数为4-5个,具有1个wt和3-4个突变等位基因(外显子组序列)。突变体rna与野生型rna的比率预测了药物诱导的p110α降解的量。在转录阶段未发生p110α的减少。图7b显示了在用他塞利昔布(gdc0032)处理的hcc1954p110α野生型(左)和p110αh1047r突变体(右)p110α细胞中相对于rpl19对照测量的p110αmrna表达的实时qpcr结果。图7c显示了在用阿哌利昔布(byl-719)处理的hcc1954p110α野生型(左)和p110αh1047r突变体(右)p110α细胞中相对于rpl19对照测量的p110αmrna表达的实时qpcr结果。野生型和突变型细胞中mrna水平的比率证实在转录阶段未发生p110α减少。
测定法名称:pik3ca.h1047r.wt
fam探针序列:atgatgcacatcatggt(seqidno.:1)
正向引物序列:ggctttggagtatttcatgaaaca(seqidno.:2)
反向引物序列:gaagatccaatccatttttgttgtc(seqidno.:3)
测定法名称:pik3ca.h1047r突变体
fam探针序列:tgatgcacgtcatggt(seqidno.:4)
正向引物序列:ggctttggagtatttcatgaaaca(seqidno.:5)
反向引物序列:gaagatccaatccatttttgttgtc(seqidno.:6)
他塞利昔布对p110α的耗竭是蛋白酶体介导的(图8a-8e),并且需要e1泛素激活酶,如图8f所示。图8a显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μm的蛋白酶体抑制剂mg132(n-(苄氧羰基)亮氨酰亮氨酰亮氨酸缩醛z-leu-leu-leu-al,cas登记号133407-82-6)(右泳道)。mg-132蛋白酶体抑制剂通过他塞利昔布(gdc-0032)挽救p110α的降解。在24小时处加入蛋白酶体抑制剂因太晚而不能防止药物诱导的降解。
p110α耗竭是蛋白酶体介导的,并且需要e1泛素激活酶。图8b显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞裂解物的western印迹分析。在收获之前4小时,加入10μm的mg132(中间泳道)和10μm的uae1抑制剂((2r,3s,4r,5r)-5-(6-(((s)-2,3-二氢-1h-茚-1-基)氨基)-9h-嘌呤-9-基)-3,4-二羟基四氢呋喃-2-基)甲基氨基磺酸盐,cas登记号905578-77-0,具有以下结构:
图8c显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132(右侧的泳道)。他塞利昔布介导p110α多聚泛素化,并且用mg132积聚多泛素化的p110α。使用e1抑制剂(uae1抑制剂)进行治疗,参见图8e,放射自显影图中的高分子量带消失(collapsed),证实了抗体的特异性。
图8d也显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132(右侧的泳道)。根据对细胞膜和胞质溶胶进行的测量的比较表明,p110α的泛素化主要发生在细胞膜上。膜相关p110α比胞质溶胶p110α更有效地被他塞利昔布泛素化。他塞利昔布迅速介导了质膜上突变型p110α的降解。膜相关p110α的降解速率比胞质溶胶p110α降解快得多。用他塞利昔布短期处理细胞介导了膜p110α而不是细胞溶质p110α的剂量依赖性降解。相比之下,阿哌利昔布(byl-719)在膜上效果较弱,对总裂解物无影响。阿哌利昔布在膜中表现出弱的初始反应,但不引起p110α随时间降解。他塞利昔布是比阿哌利昔布更优越的p110α降解剂。
图8e显示了以指示的时间用他塞利昔布1.6μm处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。在收获之前4小时,加入10μmmg132,氯喹或氯化铵(右侧的泳道)。他塞利昔布介导的p110α耗竭不受溶酶体抑制剂的影响,表明p110α降解不是核内体/溶酶体介导的,其中蛋白酶体抑制剂mg132是阳性对照。
图8f显示了p110α泛素化的途径图,描述了动物细胞通过路径特异性抑制剂降解蛋白质的两种不同机制(jadhav,t.等人(2009)“defininganembeddedcodeforproteinubiquitination”j.proteomicsbioinform,vol2(7):316-333;wang,g.等人(2012)(“k63-linkedubiquitinationinkinaseactivationandcancer”frontiersinoncology,vol2(5):1-13)。因为蛋白酶体抑制剂mg132而不是溶酶体抑制剂氯喹或氯化铵nh4cl能够挽救p110α降解,所以gdc-0032介导的p110α降解是泛素蛋白酶体依赖性降解途径,而不依赖于溶酶体降解机制。
因而,他塞利昔布介导的p110α蛋白降解是时间依赖性的,剂量依赖性的,非转录的,对突变体细胞特异的,泛素依赖性的和由蛋白酶体介导的,并且对于膜相关p110α而言是快速的。
他塞利昔布或阿哌利昔布都不影响膜上的肝脏p110α。
他塞利昔布未使p110δ降解或耗竭(图9a和9b)。图9a显示了用他塞利昔布和阿哌利昔布(byl719,novartis,cas#:1217486-61-7,(s)-n1-(4-甲基-5-(2-(1,1,1-三氟-2-甲基丙-2-基)吡啶-4-基)噻唑-2-基)吡咯烷-1,2-二甲酰胺)处理的pi3k野生型,braf突变型sw982细胞的western印迹分析。他塞利昔布未使p110δ降解或耗竭。图9b显示了用他塞利昔布和g-102(表1,us8242104)处理的pi3k野生型b细胞淋巴瘤su-dhl-10细胞的western印迹分析。
在pi3k信号通路中,pi3k抑制剂使负反馈减弱并引发再激活通路。当负反馈被阻断时,磷酸化rtk(erbb2,egfr,erbb3)活性增加,pi3k抑制的有效性降低,并且pakt增加。图10显示了以不同浓度(100nm,1μm,5μm)的pi3k抑制剂以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的pik3ca野生型hdqp1细胞的western印迹分析。
在晚期时间点维持信号传导抑制方面,他塞利昔布比另一种pi3k抑制剂g-102(us8242104)更好。图11a显示了以不同浓度(3nm,16nm,60nm,400nm,2μm)的g-102以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的mda-mb453(h1047r)细胞的western印迹分析。图11b显示了以不同浓度(3nm,16nm,60nm,400nm,2μm)的他塞利昔布(gdc-0032)以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的mda-mb453(h1047r)细胞的western印迹分析。
他塞利昔布防止rtk驱动的通路再激活。在1小时时,两种pi3k抑制剂的pakt敲低大致相同。在24小时时,prtk增加。在24小时时,他塞利昔布比g-102(一种非降解剂pi3k抑制剂)更好地抑制信号传导。图12显示了以不同浓度(1nm,10nm,100nm)的他塞利昔布(gdc-0032)和g-102以及对照(dmso溶媒)进行pi3k抑制1小时和24小时的sw48h1047r细胞的western印迹分析。
在用生长因子配体刺激的细胞中,他塞利昔布在抑制信号传导方面优于非降解剂g-102。这种效应的原理是当prtk增加时,突变型pi3kα更易于降解。prtk结合被认为使p85相对于p110α迁移,导致更有活性的p110α(alpha)。热点p110α突变可能发生在p110α和p85之间的界面上,并可使p85/p110相互作用变松。图13显示了以不同浓度(1nm,10nm,100nm)的他塞利昔布(gdc-0032)和g-102以及对照(dmso溶媒)进行pi3k抑制处理的具有和没有生长因子配体nrg的sw48pik3cah1047r细胞的western印迹分析。
测量体外细胞增殖,在24小时时在相对于野生型细胞的等基因突变型细胞中的ppras40可用于在降解测定中选择pi3k抑制剂化合物。图14a-c显示了以不同浓度的g-181,gdc-0032和g-102处理的在24小时时的在相对于野生型亲本pi3k细胞的等基因突变型(e545k,h1047r)细胞中ppras40的体外细胞增殖,以建立亲本/突变体选择性ec50值。
观察到他塞利昔布在突变型pi3kα敲入细胞中相对于泛pi3k抑制剂匹克替利昔布(gdc-0941)的细胞效力增加。图15显示了sw48等基因野生型和突变型(e545k,h1047r)细胞系的体外细胞增殖数据以及用剂量逐步增高的他塞利昔布和匹克替利昔布进行处理的绘图。
他塞利昔布在pik3ca突变型癌细胞系中具有增强的效力。图16显示了匹克替利昔布(gdc-0941),bkm120(布帕利昔布(buparlisib),novartisag,cas登记号944396-07-0),5-(2,6-二吗啉基嘧啶-4-基)-4-(三氟甲基)吡啶-2-胺),他塞利昔布(gdc-0032)和byl719(阿哌利昔布,novartiscas号1217486-61-7)在针对pik3ca突变型细胞系的4天细胞增殖(cell-titerglor,promega)测定中的功效(ic50微摩尔)的绘图。每个圆点代表不同的癌细胞系。匹克替利昔布,bkm120(布帕利昔布,novartisag,cas登记号944396-07-0),和byl719是pi3k的非降解剂。
他塞利昔布在细胞凋亡测定中具有增强的效力。图17显示了利用细胞死亡-核小体elisa检测,在72小时研究中pik3ca野生型和突变型(e545k,h1047r)细胞系的体外细胞增殖数据以及用剂量逐步增高的他塞利昔布和pi3kα选择性抑制剂gdc-0326(us8242104)和byl719进行处理的绘图。
在小鼠的pi3kα突变型异种移植物中,他塞利昔布具有比其他非降解剂药物更高的最大体内功效。在最大耐受剂量(mtd)时,他塞利昔布可引起肿瘤缩小。图18a显示了荷载包含pik3cah1047r(pi3kα)突变的hcc1954.x1乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过21天的拟合的肿瘤体积变化,所述小鼠每天一次通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80),150mg/kg的匹克替利昔布(gdc-0941)和25mg/kg的他塞利昔布(gdc-0032)。术语μl表示微升。在两种药物的最大耐受剂量时,gdc-0032比gdc-0941更有效,并诱导了肿瘤消退。因此,在pi3k突变型异种移植模型中,gdc-0032比非突变型选择性pi3kα抑制剂更有效。图18b显示了荷载包含pik3cah1047r(pi3kα)突变的hcc1954.x1乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过21天的拟合的肿瘤体积变化,所述小鼠每天一次通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80),40mg/kg的阿哌利昔布(byl-719)和15mg/kg的他塞利昔布(gdc-0032)。以gdc-0032每日口服给药持续21天导致治疗(rx)期肿瘤消退。或者,以非突变型选择性pi3kα抑制剂byl-719每日口服给药21天诱导了肿瘤停滞。因此,在pi3k突变型异种移植模型中,gdc-0032比非突变型选择性pi3kα抑制剂更有效。当与载体对照或研究开始相比时,基于小鼠体重的最小变化,用gdc-0032和byl-719进行的治疗是良好耐受的。
被称为阿哌利昔布(byl719,novartis,cas#:1217486-61-7)的化合物是pi3kα同等型的口服选择性抑制剂,其处于多种肿瘤类型的潜在治疗的临床试验中,包括与氟维司群联合用于二线激素受体阳性,her2-晚期转移性乳腺癌的iii期研究(furet,p.等人(2013)bioorg.med.chem.lett.23:3741-3748;us8227462;us8476268;us8710085)。阿哌利昔布命名为(s)-n1-(4-甲基-5-(2-(1,1,1-三氟-2-甲基丙-2-基)吡啶-4-基)噻唑-2-基)吡咯烷-1,2-二甲酰胺),并具有以下结构:
图18c显示了荷载包含pik3cae542k(pi3kα)突变的源于whim20激素受体阳性患者的乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过28天的拟合的肿瘤体积变化,所述小鼠每天一次通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80)和15mg/kg的他塞利昔布(gdc-0032)。以gdc-0032每日口服给药持续28天导致在给药已经结束之后持续的治疗期的肿瘤消退。当与载体对照或研究开始相比时,基于小鼠体重的最小变化,用gdc-0032进行的治疗是良好耐受的。
图18d显示了荷载包含pik3cah1047r(pi3kα)突变的源于hci-003激素受体阳性患者的乳腺肿瘤异种移植物的8-10只免疫受损小鼠的队列经过27天的拟合的肿瘤体积变化,所述小鼠每天一次通过100微升(μl)po(口服)给药施用载体(mct;0.5%甲基纤维素/0.2%吐温80),40mg/kg的阿哌利昔布(byl-719)以及2.5,5.0,15mg/kg的他塞利昔布(gdc-0032)。以gdc-0032每日口服给药持续27天导致在治疗(rx)期肿瘤消退呈剂量依赖性增加。或者,以非突变型选择性pi3kα抑制剂byl-719每日口服给药27天诱导了肿瘤停滞。因此,在pi3k突变型异种移植模型中,gdc-0032比非突变型选择性pi3kα抑制剂更有效。当与载体对照或研究开始相比时,基于小鼠体重的最小变化,用gdc-0032和byl-719进行的治疗是良好耐受的。
在源于包含pik3cae542k热点突变的whim20激素受体阳性患者的乳腺癌异种移植模型中评估gdc-0032的功效。以gdc-0032每日口服给药持续28天导致在给药已经结束之后持续的治疗(rx)期的肿瘤消退。当与载体对照或研究开始相比时,基于小鼠体重的最小变化,用gdc-0032进行的治疗是良好耐受的。
在源于包含pik3cah1047r热点突变的hci-003激素受体阳性患者的乳腺癌异种移植模型中评估gdc-0032的功效。以gdc-0032每日口服给药持续27天导致在治疗(rx)期肿瘤消退呈剂量依赖性增加。或者,以非突变型选择性pi3kα抑制剂byl-719每日口服给药27天诱导了肿瘤停滞。因此,在pi3k突变型异种移植模型中,gdc-0032比非突变型选择性pi3kα抑制剂更有效。当与载体对照或研究开始相比时,基于小鼠体重的最小变化,用gdc-0032和byl-719进行的治疗是良好耐受的。
在用他塞利昔布处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞中,信号传导在他塞利昔布洗脱之后迅速恢复正常,p110α水平在8-24小时后开始恢复。图19显示了用不同浓度(16nm,80nm,400nm)他塞利昔布以及对照(dmso溶媒)处理的突变型hcc1954(pik3cah1047r)乳腺癌细胞的western印迹分析。
在一个实施方案中,可以通过细胞增殖,细胞信号传导或凋亡水平来测量p110α蛋白的耗竭。
在一个实施方案中,p110α蛋白的耗竭与从患者获得的生物样品的可测量生物标志物相关联。在临床环境中可以通过proteinsimpleief(等电聚焦)技术检测p110α蛋白的耗竭。使用足够的可用患者组织,western印迹分析或质谱可以测量p110α蛋白水平。质谱可以允许探询单细胞的蛋白质组,包括泛素化的检测。nmr(核磁共振)光谱学是检测和测量p110α和p85解离或p110α降解的另一种生物物理学工具。
或者,特异性抗p110α抗体可用于免疫组织化学(ihc)或基于免疫荧光(if)的试验。
pi3k蛋白的免疫沉淀(ip)和蛋白质定位可以检测p85和p110α的解离变化,或者当治疗前后p110α被降解时,他塞利昔布可以鉴定和预测有反应的患者。
他塞利昔布在突变型等基因pi3k细胞系中的活性大于在野生型等基因pi3k细胞系中的活性。等基因pi3k突变型细胞系可以具有选自h1047r,c420r,h1047l,e542k,e545k和q546r的突变。
相对于突变型p85/p110α复合物的atp-km,他塞利昔布对p85/p110α复合物的影响不同,正如通过atp-km,pip2-km,pip2转化为pip3的速率或程度,膜定位,脂质膜亲和力,和受体酪氨酸激酶结合所测定或检测的。
相对于突变型p85/p110α复合物,他塞利昔布对野生型p85/p110α复合物构象变化的诱导不同。构象变化包括他塞利昔布与突变型p85/p110α复合物的结合相互作用,所述相互作用在他塞利昔布与野生型p85/p110α复合物之间是不存在的。
在一个实施方案中,他塞利昔布选择性地结合突变体形式的分离的pi3kα,并且与突变型pi3kα结合的ic50小于与野生型pi3kα结合的ic50。
在一个实施方案中,相比于抑制野生型形式的pi3kα等基因癌细胞系,他塞利昔布更有效地抑制突变型pi3kα等基因癌细胞系。
在一个实施方案中,他塞利昔布与突变型pi3k的α亚基结合是选择性的,其中pi3k突变选自h1047r,c420r,h1047l,e542k,e545k和q546r,并在突变型pi3kp110α同等型细胞系中具有抑制活性,如通过ic50测量的,所述抑制活性低于他塞利昔布在野生型pi3kp110α同等型细胞系中的ic50抑制活性。突变型pi3kp110α同等型选自h1047r,c420r,h1047l,e542k,e545k和q546r。
胰酶切割和p110α耗竭的质谱检测
除了检测和测量用他塞维昔布处理的生物样品中的p110α的耗竭的上述方法之外,还可以通过质谱术实现直接检测。将表达野生型和h1047r突变型p110α蛋白质的混合物的hcc1954乳腺癌细胞用在dmso中的500nm他塞利昔布(gdc-0032)处理24小时。进行p110α蛋白的免疫沉淀,通过sds-page分离捕获的蛋白质,并进行考马斯染色。将含有p110α的凝胶条带进行凝胶胰蛋白酶消化,以产生供分析的胰蛋白酶肽。基于对应的749.8282m/z(+/-10ppm)离子的定量,来自用dmso(对照)或他塞利昔布处理的细胞的胰蛋白酶肽显示出相等水平的qm(ox)ndahhggwttk肽序列(seqidno.:7)。与dmso处理的细胞相比,来自用他塞利昔布处理的细胞的胰蛋白酶肽显示出突变体特异性肽hggwttk(seqidno.:8)的丧失,其特征在于393.6983m/z(+/-10ppm)的离子。因此,突变型肿瘤特异的新胰蛋白酶肽,例如p110αh1047r,可用于证明相对于野生型形式的突变型p110α的耗竭。
野生型胰蛋白酶肽qm(ox)ndahhggwttk(seqidno.:7)突变型胰蛋白酶肽hggwttk(seqidno.:8)
图24a显示了根据实施例7的野生型pik3cahcc-1954(上)和表达pik3cahcc-1954的h1047r突变(下)的胰蛋白酶切割。
图24b显示了根据实施例7的野生型pik3cahcc-1954(左)和表达pik3cahcc-1954的h1047r突变(右)在消化和p110α(pik3ca)蛋白免疫沉淀之后的液相色谱-串联质谱(lc-ms/ms)分析。
突变体选择性
表1汇编了几种pi3k结合化合物的生物学特性。相对于泛pi3k抑制剂化合物gdc-0941(folkes等人(2008)jour.ofmed.chem.51(18):5522-5532;us7781433;us8324206),gdc-0032(ndubaku等人(2013)jour.med.chem.56(11):4597-4610;staben等人(2013)bioorg.med.chem.lett.232606-2613;wo2011/036280;us8242104;us8343955)对pi3kα突变型癌细胞更有效。表1的四种pi3k抑制剂在与pi3kα结合的生物化学效力(ki值)和针对pi3k的四种野生型1型同等型的相对效力方面不同。gdc-0326是α选择性pi3k抑制剂化合物,并与β,δ和γ同等型弱结合(us8242104)。gdc-0941是一种“泛”抑制剂,与所有四种同等型的结合相对较好。gdc-0032是“β回避”的,与α,δ和γ同等型结合较好,而与β弱结合。
表1:结合pi3k的化合物的活性
突变型pi3kα的敲入增加了他塞利昔布(gdc-0032)的细胞效能,如在sw48等基因系,pi3kα野生型(亲本)和螺旋结构域突变型e545k和激酶结构域突变型h1047r中所示,而gdc-0941未显示出这种突变体选择性效应。可以推断,gdc-0032与突变蛋白质的相互作用不同于gdc-0941。这种意想不到的结果暗示了某些有效的pi3k抑制剂而不是其他抑制剂的独特结合机制或模式。在具有激酶结构域h1047r突变的pi3kα突变型异种移植肿瘤模型hcc1954乳腺癌中,gdc-0032(缺乏gdc-0941)的突变体选择性特性使gdc-0032具有比gdc-0941更高的最大功效。以最大耐受剂量25mg/kg每日口服给药21天后,gdc-0032诱导肿瘤消退,而最大耐受剂量150mg/kg的gdc-0941引起肿瘤生长抑制。
测量在用gdc-0032,gdc-0326(us8242104)或gdc-0941处理的癌细胞系中的体外肿瘤细胞增殖。gdc-0032以低浓度诱导了pi3k突变型细胞的凋亡。gdc-0032的突变体效力增强与针对野生型pi3kα的生物化学结合效力无关。pi3k抑制剂化合物对pi3k突变型细胞系和肿瘤的效力增强可能受以下因素的影响:针对野生型p110α或野生型p110δ的物理化学或渗透性,细胞内水平,同等型选择性和绝对效力。在细胞活力测定法中,p110α选择性抑制剂gdc-0326(表1)对h1047rp110α细胞系hcc1954的效力比gdc-0032低6倍。与gdc-0326(us8242104)相比,gdc-0032对β,γ和δ同等型的α选择性较低。突变型pi3kα的敲入增加了突变体选择性gdc-0032而不增加pi3kα特异性抑制剂例如g-102的细胞效力。类似的细胞活力测定法确定,p110δ的抑制未降低pi3k突变型细胞系的活力。同样,在不同的野生型和pi3k突变型细胞中测量到可比较的gdc-0032的细胞内水平(pmol/mg)。结果表明,细胞内积累并不能解释gdc-0032的突变体效力的增加。
通过western印迹分析,进行来自用gdc-0032,gdc-0326或gdc-0941处理的突变型等基因sw48pi3kαh1047r细胞系和野生型sw48亲本细胞的放射标记裂解物的凝胶电泳放射自显影,测量了切割的parp,ps6(s235/236),paktt308,pakts473,β-肌动蛋白和gapdh。pi3k通路敲低以剂量依赖性方式与诱导凋亡相关。在很低的化合物浓度下,他塞利昔布诱导包含pi3k突变的细胞的凋亡。在来自mcf10a乳腺细胞系以及hcc1954,pi3kαh1047r乳腺癌细胞系的等基因细胞中,观察到相似的效应。尽管化合物性质相当,但相比于非突变体选择性gdc-0326,具有突变体选择性gdc-0032的通路敲低更强。gdc-0032的突变体效力增强不能通过针对野生型pi3kα的生物化学效力得以解释。
通过这些测定法可以评定化合物,以检查结构变化对pi3kα突变体选择性的影响。特定区域的大小和氢键能力的变化可能与选择性提高相关。
初步临床试验数据显示,他塞利昔布在10例pik3ca突变型肿瘤患者中有5例并且在5例pik3ca突变型乳腺肿瘤患者中有4例达到部分缓解(olivero和juric(2013)aacr)。
他塞利昔布与pi3k的相互作用
由于与pi3kα突变体同等型的关键相互作用不同于与pi3k野生型同等型的相互作用,他塞利昔布(gdc-0032)对pi3kα突变体同等型相比于对pi3k野生型同等型更有选择性,并且可包括原子和官能团的精确定位和排列,以与pi3kα突变体同等型的关键突变体特异性特征相互作用。这样的相互作用可以通过用作氢键供体,氢键受体的官能团和/或具有pi3kα突变体同等型蛋白质的范德华力配偶体来实现(staben等人(2013)bioorg.med.chem.lett.23:2606-2613;ndubaku等人(2013)jour.med.chem.56(11):4597-4610)。他塞利昔布可以采用低能构象的结合拓扑结构,并在配体结合位点中产生有效的极性相互作用和范德华相互作用。
已经确定,pik3ca突变增加了脂质结合和pi3k基础活性(burke等人(2012)proc.natl.acad.sci.109:15259-15264)。突变使封闭的胞质无活性形式的pi3kα不稳定,从而促进脂质结合。本发明的突变体选择性pi3k结合化合物增加了封闭形式的pi3kα的稳定性,从而阻止使脂质结合增加的构象变化。在h1047r突变体的氢氘交换中,热点突变诱导了更多的激酶结构域区域的氘化,氘化表明所述区域变得更加动态(不稳定)并可用于交换。这些变化伴随着对脂质膜的亲和力增加,并且可以解释活化和下游信号传导的增加。这个动态区域,残基848-859(图20),可能为本发明化合物提供关键的结合相互作用。
图21显示了根据pik3ca中突变位置,在具有pik3ca突变体的细胞系中gdc-0032活力的结合效力绘图。gdc-0032对于包含pi3k突变的细胞系是有效的,无论突变的位置如何。一些细胞系具有另外的突变,例如作为抗性标记的b-raf和ras。
图22显示了p85与p110α的免疫共沉淀(co-ip)的western印迹(wb)分析,表明稳定的p110α与p85复合的与p110α平行的水平以及由他塞利昔布诱导的显著剂量依赖性p110α降解。细胞用gdc-0032处理24小时,在该点时p110α明显降解。替代实验可以采用在较短时间点(2小时,4小时等)取样的细胞处理,其中p110α尚未降解,但与p85解离。检测用他塞利昔布处理后突变型而不是野生型p110α与p85的解离,其可以是突变型pi3k肿瘤患者可能对他塞利昔布治疗作出响应的预测性生物标志物。
图23显示了稳态p110αmrna表达。对gdc0032使p110α降解最敏感的细胞系比wtp110α包含更多的h1047r突变。突变型与wtp110α等位基因的比率可能决定对gdc-0032介导的p110α降解的敏感性。
已经报道了pi3kβ(zhang,x等人(2011)mol.cell41:567–5789),pi3kα(huang,c.-h.等人(2007)science318:1744),pi3kδ(berndt,a.等人(2010)nat.chem.biol.6:11)和pi3kγ("structuraldeterminantsofphosphoinositide3-kinaseinhibitionbywortmannin,ly294002,quercetin,myricetin,andstaurosporine",walker,e.h.etal(2000)mol.cell6:909)的x射线结构。一个值得注意的区别在于所有同等型(α-trp780,β-trp781,δ-trp760,γ-trp812)中存在的色氨酸残基的构象。由于相邻残基的“诱导拟合”运动或抑制剂的特异性相互作用,这是被认为对于获得pi3kδ的同等型特异性关键的相同色氨酸残基。该trp残基的吲哚的构象在pi3kβ中是独特的,并且可以为与pi3kβ低,弱结合的“β回避”型pi3k抑制剂的设计提供依据。β-trp781沿着cα-cβ的旋转呈现出另一个取向。第二壳残基的差异促进了pi3kβ中的这个trp的独特取向。首先,吲哚n-h向pi3kα(glu798)和pi3kγ(glu814)二者中的酸性残基提供h键,所述酸性残基占据结合位点的相似区域,但在pi3kβ或δ中不存在。这种相互作用可能有利于观察到的吲哚在这些同等型中的取向。代替glu814,pi3kβ和δ具有带非极性侧链的中性残基(分别为β-val783和δ-met762),因此吲哚可以占据其他在能量上有利的取向。pi3kβ的支链缬氨酸残基可能不利于与在α,δ和γ中观察到的取向相似的取向;而cγ甲基改为接近吲哚4/5位置的范德华距离。在这种结构-活性关系(sar)的解释下,在这种独特的构象中,trp侧链更容易受到抑制剂的空间体积的刺激。还值得注意的是通过相对于pi3kβ的pi3kα,δ和γ中的吲哚的π堆叠所呈现的差角和原子。对于色氨酸环和芳香族氨基酸侧链(phe,tyr,his)之间的π相互作用的pdb分析参见:samanta,u.等人(1999)biol.crystallogr.,d55:1421。这一观察能够部分地解释为什么在相同区域中的非芳香族取代导致在pi3kb上具有更低的总选择性的分子(staben等人(2013)bioorg.med.chem.lett.23:2606–2613)。
突变型p110α的生物标志物检测
在某些实施方案中,使用免疫组织化学(ihc)和染色方案检查样品中生物标志物蛋白的存在和/或表达水平/量。已经显示组织切片的ihc染色是确定或检测样品中蛋白质存在的可靠方法。在任何方法,测定法和/或试剂盒的一些实施方案中,相关的生物标志物是突变型p110α蛋白。在一些实施方案中,通过免疫组织化学检测突变型p110α。在一些实施方案中,在来自个体的样品中的突变型p110α生物标志物的表达升高为蛋白质表达升高,并且在进一步的实施方案中是使用ihc确定的。在一个实施方案中,使用以下方法确定生物标志物的表达水平,所述方法包括:(a)用抗体对样品(比如受试者癌症样品)进行ihc分析;和b)确定样品中生物标志物的表达水平。在一些实施方案中,相对于参考确定ihc染色强度。在一些实施方案中,所述参考是参考值。在一些实施方案中,所述参考是参考样品(例如,来自非癌症患者的对照细胞系染色样品或组织样品)。
ihc可以与另外的技术联合进行,例如形态染色和/或荧光原位杂交。可用ihc的两种一般方法;直接测定法和间接测定法。根据第一种测定法,直接确定抗体与靶抗原的结合。这种直接测定法使用标记的试剂,例如荧光标签或酶标记的一级抗体,其可以在没有进一步的抗体相互作用的情况下被可视化。在典型的间接测定法中,未缀合的一级抗体与抗原结合,然后标记的二级抗体与所述一级抗体结合。当二级抗体与酶标记物缀合时,加入生色或荧光底物来提供抗原的可视化。由于若干二级抗体可与一级抗体上的不同表位反应,发生信号扩增。用于ihc的一级和/或二级抗体通常用可检测模块加以标记。多种标记物可供使用,通常可以分为以下类别:(a)放射性同位素,如35s,14c,125i,3h和131i;(b)胶体金颗粒;(c)荧光标记物,包括但不限于稀土螯合物(铕螯合物),德克萨斯红,罗丹明,荧光素,丹磺酰,丽丝胺,伞形酮,藻红蛋白(phycocrytherin),藻蓝蛋白或市售的荧光团如spectrumorange7和spectrumgreen7和/或上述任何一者或多者的衍生物;(d)各种酶底物标记可供使用(us4275149;us4318980)。酶标记物的实例包括荧光素酶(例如,萤火虫荧光素酶和细菌荧光素酶(us4737456)),荧光素,2,3-二氢酞嗪二酮,苹果酸脱氢酶,脲酶,过氧化物酶如辣根过氧化物酶(hrpo),碱性磷酸酶,β-半乳糖苷酶,葡糖淀粉酶,溶菌酶,糖氧化酶(例如,葡萄糖氧化酶,半乳糖氧化酶和葡萄糖-6-磷酸脱氢酶),杂环氧化酶(例如尿酸酶和黄嘌呤氧化酶),乳过氧化物酶,微过氧化物酶等。酶-底物组合的实例包括例如,辣根过氧化物酶(hrpo)与作为底物的过氧化氢酶;碱性磷酸酶(ap)与作为生色底物的对硝基磷酸苯酯;和β-d-半乳糖苷酶(β-d-gal)与生色底物(例如,对硝基苯基-β-d-半乳糖苷酶)或荧光底物(例如,4-甲基伞形酮基-β-d-半乳糖苷酶)。在任何方法的一些实施方案中,使用抗突变型p110α诊断抗体(即,一级抗体)通过免疫组织化学检测诸如突变型p110α蛋白的生物标志物。在一些实施方案中,抗突变型p110α诊断抗体特异性结合人突变型p110α。在一些实施方案中,抗突变型p110α抗体是非人抗体,例如大鼠,小鼠或兔抗体。在一些实施方案中,抗突变型p110α诊断抗体是单克隆抗体。在一些实施方案中,抗突变型p110α诊断抗体被直接标记。
如此制备的标本可以被封固和加盖玻片。然后例如使用显微镜确定玻片评估,并且可以采用本领域常规使用的染色强度标准。在一个实施方案中,应当理解,当使用ihc检查来自肿瘤的细胞和/或组织时,通常确定或评定肿瘤细胞和/或组织中的染色(与可能存在于样品中的基质或周围组织相反)。在一些实施方案中,应当理解,当使用ihc检查来自肿瘤的细胞和/或组织时,染色包括确定或评定包括瘤内或肿瘤周围免疫细胞在内的肿瘤浸润性免疫细胞。在一些实施方案中,通过在>0%的样品中,在至少1%的样品中,在至少5%的样品中,在至少10%的样品中的ihc,检测突变型p110α生物标志物的存在。在一些实施方案中,通过肿瘤细胞中的ihc检测突变型p110α生物标志物。
在替代方法中,可以使来自患者的生物样品,例如肿瘤活检或循环肿瘤细胞在足以形成抗体-生物标志物复合物的条件下与生物标志物特异的抗体接触,然后检测所述复合物。可以多种方式检测生物标志物的存在,例如通过用于测定各种组织和样品(包括血浆或血清)的western印迹和elisa程序。使用这种测定形式的各种免疫测定技术是可用的(us4016043;us4424279;us4018653),包括非竞争性类型的单位点和双位点或“夹心”测定法,以及传统的竞争性结合测定法。这些测定法还包括使标记的抗体与靶标生物标志物直接结合。还可以通过功能性或基于活性的测定法来检查组织或细胞样品中所选生物标志物的存在和/或表达水平/量。例如,如果生物标志物是酶,则可以进行本领域已知的测定法来确定或检测组织或细胞样品中给定酶活性的存在。
可以使用本领域已知的方法制备抗突变型p110α抗体或其抗原结合片段,例如通过这样的过程,其包括:在适合于产生此类抗体或片段的条件下培养含有编码任何前述抗突变型p110α抗体或抗原结合片段的核酸的宿主细胞,和回收所述抗体或片段。
治疗方法
他塞利昔布(gdc-0032)可用于治疗包括癌症在内的过度增殖性疾病。在一个实施方案中,用他塞利昔布治疗患有突变型pi3k肿瘤的患者。突变型pi3k肿瘤可能是乳腺肿瘤,肺肿瘤或其他器官中发现的肿瘤。
本发明的一个实施方案是治疗癌症的方法,其包括向患者施用他塞利昔布,其中他塞利昔布在突变型等基因pi3k细胞系中的活性大于他塞利昔布在野生型等基因pi3k细胞系中的活性。
本发明的另一个实施方案是治疗癌症的方法,其包括向患者施用他塞利昔布,其中他塞利昔布与突变型pi3kp110α同等型的ic50结合活性低于他塞利昔布与野生型pi3kp110α同等型的ic50结合活性。
在一个实施方案中,pi3kp110α同等型突变体选自h1047r,h1047l,e542k,e545k和q546r。
在一个实施方案中,已经测试了在施用他塞利昔布之前从患者获得的生物样品的pik3ca或pten突变状态,并且其中pik3ca或pten突变状态指示患者对他塞利昔布的治疗响应性。
在一个实施方案中,突变状态包括检测选自h1047r,h1047l,c420r,e542k,e545k或q546r的突变体。
在一个实施方案中,过度增殖性疾病是表达her2的乳腺癌或雌激素受体阳性(er+)乳腺癌,其中乳腺癌可以是转移性的。
在一个实施方案中,以辅助治疗的形式向患者施用他塞利昔布。
在一个实施方案中,患者先前已经接受用他莫西芬,氟维司群或来曲唑治疗。
本发明的方法还包括:
·基于生物标志物鉴定的诊断方法;
·确定患者是否会对他塞利昔布或他塞利昔布与化疗剂的联合作出响应的方法;
·通过监测他塞利昔布或他塞利昔布与化疗剂的联合的清除率来优化治疗效果的方法;
·通过监测治疗性抗性突变的发展,优化他塞利昔布或他塞利昔布与化疗剂的联合的治疗方案的方法;和
·确定哪些患者将会最受益于他塞利昔布或他塞利昔布与化疗剂疗法联合进行治疗的方法,以及监测所述患者对用他塞利昔布或他塞利昔布与化疗剂疗法联合进行治疗的敏感性和响应性的方法。
本发明的方法可用于抑制异常细胞生长或治疗哺乳动物(例如人类患者)中的过度增殖性疾病,如癌症。例如,所述方法可用于诊断,监测和治疗哺乳动物(例如人)中的多发性骨髓瘤,淋巴瘤,白血病,前列腺癌,乳腺癌,肝细胞癌,胰腺癌和/或结肠直肠癌。
(1)他塞利昔布和(2)化疗剂的治疗联合可用于治疗包括但不限于以pi3激酶途径激活为特征的那些疾病,病症和/或失调。因此,本发明的另一方面包括治疗可通过抑制脂质激酶(包括pi3k)进行治疗的疾病或病症的方法。在一个实施方案中,治疗实体瘤或造血系统恶性肿瘤的方法包括:向哺乳动物施用包括他塞利昔布作为联合制剂的治疗联合或交替地进行给药,其中治疗联合包括治疗有效量的他塞利昔布,以及治疗有效量的选自5-fu,多西他赛,艾日布林,吉西他滨,考比替尼(gdc-0973,xl-518,cas登记号934660-93-2),gdc-0623(cas登记号1168091-68-6),帕利他赛,他莫昔芬,氟维司群,地塞米松,帕妥珠单抗,曲妥珠单抗-艾美坦辛,曲妥珠单抗和来曲唑的一种或多种化疗剂。(1)他塞利昔布和(2)化疗剂的治疗联合可用于治疗过度增殖性疾病或失调,包括造血系统恶性肿瘤,肿瘤,癌症和肿瘤组织,以及恶性肿瘤前和非肿瘤性或非恶性过度增殖性疾病。在一个实施方案中,用治疗组合和药学上可接受的载体,佐剂或溶媒处理人类患者,其中所述治疗组合的他塞利昔布,或其代谢物以可检测地抑制pi3激酶活性的量存在。
造血系统恶性肿瘤包括非霍奇金淋巴瘤,弥漫性大造血淋巴瘤,滤泡性淋巴瘤,套细胞淋巴瘤,慢性淋巴细胞白血病,多发性骨髓瘤,aml和mcl。
本发明的另一方面提供了用于治疗本文所述的在哺乳动物中的疾病或病症的药物组合物或治疗组合,所述哺乳动物例如患有这种疾病或病症的人。还提供了药物组合物在制备用于治疗本文所述的在温血动物中的疾病和病症的药剂中的用途,所述温血动物如患有这种失调的哺乳动物,例如人。
本发明的另一方面提供了用于治疗癌症的他塞利昔布,其中待治疗的患者具有pik3ca突变,其中该突变包括选自h1047r,c420r,h1047l,e542k,e545k和q546r的突变。
本发明的另一方面提供了用于治疗癌症的他塞利昔布,其中待治疗的患者具有pik3ca突变,并且在他塞利昔布与具有pik3ca突变状态的生物样品接触之后p110α同等型被耗竭,其中该突变包括选自h1047r,c420r,h1047l,e542k,e545k和q546r的突变。
本发明的另一方面提供了他塞利昔布在制造用于治疗癌症的药剂中的用途,其中待治疗的受试者具有pik3ca突变。
本发明的另一方面提供了他塞利昔布在制造用于治疗癌症的药剂中的用途,其中待治疗的患者具有pik3ca突变,并且在他塞利昔布与具有pik3ca突变状态的生物样品接触之后p110α同等型被耗竭,其中该突变包括选自h1047r,c420r,h1047l,e542k,e545k和q546r的突变。
本发明的另一方面提供了用于治疗癌症的他塞利昔布,其中患者在接触他塞利昔布之后p110α同等型被耗竭。
本发明的另一方面提供了确定对他塞利昔布的响应性的方法,该方法包括以下步骤:
a)施用他塞利昔布;和
b)测定从所述患者获得的生物样品中的p110α水平或与p110α水平相关的生物标志物的水平的变化。
本发明的另一方面提供了如上所述的发明。
药物组合物和制剂
本发明化合物的药物组合物或制剂包含他塞利昔布和一种或多种药学上可接受的载体,助流剂,稀释剂和赋形剂。
本发明化合物的药物组合物或制剂进一步包含第二化疗剂。
本发明的突变体选择性的,结合pi3k的化合物和化疗剂可以非溶剂化形式和与药学上可接受的溶剂如水,乙醇等一起以溶剂化形式存在,并且本发明意在包括溶剂化和非溶剂化形式二者。
本发明的化合物也可以以不同的互变异构形式存在,并且所有这样的形式都被包括在本发明的范围内。术语“互变异构体”或“互变异构形式”是指不同能量的可通过低能量屏障相互转换的结构异构体。例如,质子互变异构体(也称为质子转移互变异构体)包括通过质子迁移的相互转换,例如酮-烯醇和亚胺-烯胺异构化。价键互变异构体包括通过一些成键电子的重组的相互转换。
药物组合物包括本体组合物和单独剂量单位两者,其由多于一种(例如两种)药物活性剂以及任何药学上无活性的赋形剂,稀释剂,载体或助流剂组成,所述药物活性剂包括突变体选择性的,结合pi3k的化合物以及选自本文所述的另外的药剂的清单的化疗剂。本体组合物和每个单独剂量单位可以含有固定量的上述药物活性剂。本体组合物是尚未形成单独剂量单位的材料。说明性的剂量单位是口服剂量单位,比如片剂,丸剂,胶囊剂等等。类似地,通过施用药物组合物治疗患者的方法也意在涵盖施用本体组合物和单独剂量单位。
药物组合物还包括与本文列举相同的本发明的同位素标记化合物,但是实际上一个或多个原子被具有与通常在自然界中发现的原子质量或质量数不同的原子质量或质量数的原子替代。如所指定的任何特定原子或元素的所有同位素预期均落入本发明化合物及其用途的范围内。可纳入本发明化合物的示例性同位素包括氢,碳,氮,氧,磷,硫,氟,氯和碘的同位素,如2h,3h,11c,13c,14c,13n,15n,15o,17o,18o,32p,33p,35s,18f,36cl,123i和125i。本发明的某些同位素标记化合物(例如,用3h和14c标记的那些化合物)可用于化合物和/或底物组织分布测定法中。氚化(3h)和碳14(14c)同位素对于其制备和可检测性的易化是有用的。此外,用较重的同位素如氘(2h)取代可能由于更好的代谢稳定性而提供某些治疗优点(例如,体内半衰期增加或剂量要求降低),因此在一些情况下可能是优选的。正电子发射同位素如15o,13n,11c和18f可用于正电子发射断层摄影术(pet)研究中,以检查底物受体占有率。通过用同位素标记试剂取代非同位素标记试剂,本发明的同位素标记化合物通常可以通过类似于下文实施例中公开的下列程序进行制备。
根据在包括人在内的哺乳动物中的过度增殖性疾病的治疗性治疗(包括预防性治疗)的治疗联合中使用的标准药物实践来配制他塞依昔布。本发明提供了包含与一种或多种药学上可接受的载体,助流剂,稀释剂,添加剂或赋形剂结合的突变体选择性的,结合pi3k的化合物的药物组合物。
适合的载体,稀释剂,添加剂和赋形剂是本领域技术人员熟知的,包括碳水化合物,蜡,水溶性和/或可溶胀聚合物,亲水或疏水材料,明胶,油,溶剂,水等材料。使用的具体载体,稀释剂或赋形剂将取决于施用本发明化合物的手段和目的。通常基于本领域技术人员认可的对哺乳动物施用安全(gras)的溶剂来选择溶剂。一般而言,安全溶剂是无毒的水性溶剂,比如水和可溶于水或混溶于水的其他无毒溶剂。适合的水性溶剂包括水,乙醇,丙二醇,聚乙二醇(例如peg400,peg300),二甲亚砜(dmso),克列莫佛(例如cremophor
可以针对各种给药途径和类型制备本发明化合物的药物制剂。例如,具有期望纯度的突变体选择性的结合pi3k的化合物可以任选地以冻干制剂,研磨粉末或水溶液的形式与药学上可接受的稀释剂,载体,赋形剂或稳定剂混合(remington′spharmaceuticalsciences(1995)18thedition,mackpubl.co.,easton,pa)。
将以符合良好医学实践的方式(即,量,浓度,时间表,疗程,载体和给药途径)进行本发明的药物制剂的给药和施用。
他塞利昔布每剂量口服或肠胃外给药的初始药学有效量将在约0.01-1000mg/kg的范围内,即每天约0.1至20mg/kg患者体重,其中所使用的化合物的一般初始范围为0.3至15mg/kg/天。有待施用的他塞利昔布的剂量和化疗剂的剂量的范围可以各自为每单位剂型约1mg至约1000mg,或每单位剂型约10mg至约100mg。可以按重量计以约1:50至约50:1的比率或以约1:10至约10:1的比率施用他塞利昔布和化疗剂的剂量。
处于采用剂量和浓度的可接受的稀释剂,载体,赋形剂和稳定剂对于接受者是无毒的,并且包括诸如磷酸盐,柠檬酸盐,和其他有机酸的缓冲剂;包括抗坏血酸和甲硫氨酸在内的抗氧化剂;防腐剂(如十八烷基二甲基苄基氯化铵;氯化六甲双铵;苯扎氯铵,苄索氯铵;苯酚,丁醇或苄醇;烷基对羟基苯甲酸酯,如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯;儿茶酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(少于约10个残基)多肽;蛋白质,如血清白蛋白,明胶,或免疫球蛋白;亲水性聚合物,如聚乙烯吡咯烷酮;氨基酸,如甘氨酸,谷氨酰胺,天冬酰胺,组氨酸,精氨酸,或赖氨酸;单糖类,二糖类,和其他碳水化合物类,包括葡萄糖,甘露糖,或糊精;螯合剂,如edta;糖类,如蔗糖,甘露醇,海藻糖或山梨糖醇;成盐反离子,如钠;金属复合物(例如,zn-蛋白质复合物);和/或非离子型表面活性剂,如吐温
可以制备他塞利昔布和化疗化合物的缓释制剂。缓释制剂的适合实例包括含有他塞利昔布的固体疏水聚合物的半透性基质,所述基质呈成形物品的形式,例如薄膜,或微胶囊。缓释基质的实例包括聚酯,水凝胶(例如,聚(2-羟乙基-甲基丙烯酸酯),或聚(乙烯醇)),聚丙交酯(us3773919),l-谷氨酸和γ-乙基-l-谷氨酸盐的共聚物,不可降解的乙烯乙酸乙烯酯,可降解的乳酸-乙醇酸共聚物,如lupron
药物制剂包括适合于本文详述的给药途径的那些药物制剂。这些制剂可以被合宜地提供为单位剂型,并且可以通过药学领域中熟知的任何方法进行制备。技术和制剂通常发现于remington′spharmaceuticalsciences18thed.(1995)mackpublishingco.,easton,pa。这样的方法包括使活性成分与构成一种或多种辅助成分的载体结合的步骤。一般而言,所述制剂通过以下步骤进行制备:使活性成分与液体载体或精细分散的固体载体或它们两者均匀地且精细地结合,并且如果需要的话,进而将产品成形。
适合于口服给药的他塞利昔布和/或化疗剂的制剂可以制备成离散单位,比如丸剂,(例如硬或软的)明胶胶囊,扁囊剂,糖锭,锭剂,水性或油悬浮液,可分散粉末或颗粒,乳剂,糖浆或酏剂,其各自含有预定量的他塞利昔布和/或化疗剂。他塞利昔布的量和化疗剂的量可以作为联合制剂配制在丸剂,胶囊,溶液或悬浮液中。或者,他塞利昔布和化疗剂可以分开配制在丸剂,胶囊,溶液或悬浮液中以交替方式给药。
可以根据本领域已知的制造药物组合物的任何方法制备所述制剂,并且这样的组合物可以含有一种或多种作用剂,包括甜味剂,调味剂,着色剂和防腐剂,以提供适口的制剂。可以通过在合适的机器中将自由流动形式的活性成分如粉末或颗粒压制来制备压制片剂,所述活性成分任选地与粘合剂,润滑剂,惰性稀释剂,防腐剂,表面活性剂或分散剂混合。可以通过在合适的机器中模制用惰性液体稀释剂润湿的粉末化活性成分的混合物来制备模制片剂。可任选地将片剂包衣或刻痕,并且任选配制成提供活性成分从其中缓慢释放或控制释放。
本发明药物制剂的片剂赋形剂可以包括:增加构成片剂的粉末化药物的总体积的填充剂(或稀释剂);在摄入时促进片剂分解成小碎片(理想的是单独的药物颗粒)并促进药物快速溶解和吸收的崩解剂;确保颗粒和片剂能够以所需的机械强度形成,并在压片后将片剂保持在一起,防止在包装,运输和常规处理过程中分解成其组分粉末的粘合剂;改善生产过程中组成片剂的粉末的流动性的助流剂;确保制造过程中压片粉末不粘附于用来压片的设备上的润滑剂。它们通过加压改善粉末混合物的流动,并使成品片剂从设备中射出时的摩擦力和破裂最小化;具有类似于助流剂的功能的抗粘附剂,其降低组成片剂的粉末与用于在制造过程中冲压出片剂形状的机器之间的粘附;掺入片剂中使其具有更愉快的味道或掩盖不愉快的味道的调味剂,以及有助于识别和患者依从性的着色剂。
含有与适合于制造片剂的无毒的药学上可接受的赋形剂混合的活性成分的片剂是可接受的。这些赋形剂可以是,例如惰性稀释剂,如碳酸钙或碳酸钠,乳糖,磷酸钙或磷酸钠;造粒崩解剂,如玉米淀粉或海藻酸;粘合剂,如淀粉,明胶或阿拉伯胶;和润滑剂,如硬脂酸镁,硬脂酸或滑石。片剂可以是未包衣的,或者可以通过已知的技术进行包衣,所述技术包括微囊化以延缓胃肠道中的崩解和吸附,从而在更长的时间内提供持续的作用。例如,可以单独使用诸如单硬脂酸甘油酯或二硬脂酸甘油酯等的延时材料,或与蜡一起使用。
药物组合物可以呈无菌注射制剂的形式,例如无菌注射的水性或油性悬浮液。可以使用上述适合的分散剂或润湿剂和悬浮剂根据已知技术配制这种悬浮液。无菌注射制剂可以是在无毒的肠胃外可接受的稀释剂或溶剂中的溶液或悬浮液,例如在1,3-丁二醇中的溶液或由冻干粉末制备的溶液。可以使用的可接受的溶媒和溶剂包括水,林格氏溶液和等渗氯化钠溶液。另外,无菌,不挥发性油通常可用作溶剂或悬浮介质。为了这个目的,任何无刺激性的不挥发性油都可以使用,包括合成的单甘油酯或甘油二酯。另外,脂肪酸如油酸也可以用于制备注射剂。
制剂可以包装在单位剂量或多剂量容器例如密封的安瓿和小瓶中,并且可以在冷冻干燥(冻干)条件下储存,在使用之前仅需要立即加入用于注射的无菌液体载体,例如水。由前述类型的无菌粉末,颗粒和片剂制备临时注射溶液和悬浮液。优选的单位剂量制剂是含有活性成分的如上所述的日剂量或单位每日亚剂量或其适当部分的那些制剂。
本发明进一步提供了包含至少一种上述活性成分的兽药组合物,因此将所述活性成分与兽药载体一起定义。兽药载体是用于为了施用组合物的材料,并且可以是在其他方面惰性的或在兽医学领域中可接受的并且与活性成分相容的固体,液体或气体材料。可以肠胃外,口服或任何其他期望的途径施用这些兽药组合物。
联合治疗
他塞利昔布可以与某些化疗剂联合使用,用于治疗包括实体瘤或造血系统恶性肿瘤在内的过度增殖性疾病,以及恶性肿瘤前和非肿瘤性或非恶性过度增殖性疾病。在某些实施方案中,他塞利昔布与化疗剂联合成为单一制剂,如单一片剂,丸剂,胶囊或溶液,以便同时施用所述联合。在其他实施方案中,根据给药方案或疗程以分开的制剂施用他塞利昔布和化疗剂,所述分开的制剂如用于依次施用他塞利昔布和化疗剂的分开的片剂,丸剂,胶囊或溶液,所述化疗剂选自5-fu,多西他赛,艾日布林,吉西他滨,gdc-0973,gdc-0623,帕利他赛,他莫昔芬,氟维司群,地塞米松,帕妥珠单抗,曲妥珠单抗-艾美坦辛,曲妥珠单抗和来曲唑。化疗剂具有抗过度增殖性质,或用于治疗过度增殖性疾病。他塞利昔布与化疗剂的联合可具有协同性。药物联合制剂或给药方案中的化疗剂优选地与他塞利昔布具有互补活性,并且使它们不会不良地彼此影响。可以按照对于预期目的有效的量施用治疗联合中的这些化合物。在一个实施方案中,本发明的药物制剂包含他塞利昔布和如本文所述的化疗剂。在另一个实施方案中,通过给药方案施用治疗联合,其中以每日2次到每3周1次(q3wk)的范围施用治疗有效量的他塞利昔布,并且以交替方式以每日2次到每3周1次的范围分开施用治疗有效量的化疗剂。
本发明的治疗联合包括他塞利昔布,以及选自5-fu,多西他赛,艾日布林,吉西他滨,gdc-0973,gdc-0623,帕利他赛,他莫昔芬,氟维司群,地塞米松,帕妥珠单抗,曲妥珠单抗-艾美坦辛,曲妥珠单抗和来曲唑的化疗剂,用于在过度增殖性疾病的治疗中分开,同时或依次使用。
可以按照同时方案或依次方案施用联合治疗。当依次施用时,可以按照两次或更多次给药来施用所述联合。联合给药包括共同给药,使用分开的制剂或单一药物制剂,以及以任一顺序连续给药,其中优选地存在两种(或全部)活性剂同时发挥其生物活性的时间段。
任何上述共同给药的作用剂的适合剂量是目前使用的那些剂量,并且可能由于新鉴定的作用剂和其他化疗剂或治疗的联合作用(协同作用)而降低,从而增加治疗指数或减轻毒性或其他副作用或后果。
在抗癌治疗的具体实施方案中,治疗联合可以与作为辅助治疗的外科治疗和放射治疗联合。根据本发明的联合治疗包括他塞利昔布和一种或多种其他癌症治疗方法或方式的施用。将选择他塞利昔布和化疗剂的量和给药的相对时间,以达到期望的联合治疗效果。
药物组合物的施用
可以通过适合于待治疗病症的任何途径施用本发明的治疗联合。适合的途径包括口服,肠胃外(包括皮下,肌内,静脉内,动脉内,吸入,皮内,鞘内,硬膜外和输注技术),透皮,直肠,鼻,局部(包括颊和舌下),阴道,腹膜内,肺内和鼻内。局部给药还可以涉及使用透皮给药,如透皮贴剂或离子电渗装置。药物的制剂论述于remington′spharmaceuticalsciences,18thed.,(1995)mackpublishingco.,easton,pa。药物制剂的其他实例可见于liberman,h.a.和lachman,l.,eds.,pharmaceuticaldosageforms,marceldecker,vol3,2nded.,newyork,ny。对于局部免疫抑制治疗,可以通过病变内给药施用化合物,包括在移植前灌注或以其他方式使移植物与抑制剂接触。应当理解,优选的途径可以随着例如接受者的状况而变化。当口服施用化合物时,其可以与药学上可接受的载体,助流剂或赋形剂一起配制成丸剂,胶囊,片剂等。当肠胃外施用化合物时,其可以与药学上可接受的肠胃外溶媒或稀释剂一起配制,并且被配制为单位剂量注射形式,如下详述。
治疗人类患者的剂量范围可以为约1mg至约1000mg的他塞利昔布,比如约5mg至约20mg的该化合物。取决于药代动力学(pk)和药效学(pd)性质,包括特定化合物的吸收,分布,代谢和排泄,可以每天一次(qd),每天两次(bid)或更频繁地施用剂量。另外,毒性因素可能影响剂量和给药剂量方案。当口服给药时,可以每天两次,每天一次或更低的频率,如每周一次或每两周或每三周一次摄入丸剂,胶囊或片剂,持续规定的时间段。该方案可以重复多个治疗循环。
制品
在本发明的另一个实施方案中,提供了可用于治疗上述疾病和失调的含有他塞利昔布的制品或“试剂盒”。在一个实施方案中,该试剂盒包括容纳他塞利昔布的容器。该试剂盒进一步包括在容器上或与容器相关联的标签或包装说明书。术语“包装说明书”用于指通常包括在治疗产品的商业包装中的说明书,其包含关于使用这种治疗产品的适应症,用法,剂量,给药,禁忌症和/或警告的信息。适合的容器包括例如瓶,小瓶,注射器,泡罩包装等。可以由各种材料如玻璃或塑料形成所述容器。所述容器可以容纳有效治疗病症的他塞利昔布或其制剂,并且可以具有无菌进入口(例如,该容器可以是静脉内溶液袋或具有可被皮下注射针可刺穿的塞子的小瓶)。组合物中的至少一种活性剂是他塞利昔布。标签或包装说明书指示该组合物用于治疗选择的病症,比如癌症。在一个实施方案中,标签或包装说明书指示包含式i化合物的组合物可用于治疗由异常细胞生长引起的失调。所述标签或包装说明书还可以指示该组合物可用于治疗其他失调。替代地或另外地,制品可以进一步包括第二容器,其包含药学上可接受的缓冲剂,例如抑菌性注射用水(bwfi),磷酸盐缓冲盐水,林格氏溶液和右旋糖溶液。它还可以包括立足于商业和用户立场所需的其他物品,包括其他缓冲剂,稀释剂,过滤器,针头和注射器。
所述试剂盒可以进一步包括施用他塞利昔布以及(如果存在的话)第二药物制剂的说明。例如,如果该试剂盒包括包含他塞利昔布的第一组合物和第二药物制剂,则该试剂盒可以进一步包括用于向有需要的患者同时,依次或分开施用第一和第二药物组合物的说明。
在另一个实施方案中,所述试剂盒适用于递送他塞利昔布的固体口服形式,例如片剂或胶囊。这样的试剂盒优选包括多个单位剂量。这样的试剂盒可以包括具有剂量的卡片,所述剂量按照其预期使用的顺序定向。这样的试剂盒的实例是“泡罩包装”。泡罩包装在包装行业中是熟知的,并且广泛用于包装药物单位剂型。如果需要的话,可以提供记忆辅助手段,例如呈数字,字母或其他标记的形式或日历插入物,标明在治疗计划中可以施用所述剂量的日期。
根据一个实施方案,试剂盒可以包括:(a)其中容纳他塞利昔布的第一容器;和任选地(b)其中容纳第二药物制剂的第二容器,其中所述第二药物制剂包含具有抗过度增殖活性的第二化合物。替代地或另外地,所述试剂盒可以进一步包括第三容器,其包含药学上可接受的缓冲剂,例如抑菌性注射用水(bwfi),磷酸盐缓冲盐水,林格氏溶液和右旋糖溶液。它还可以包括立足于商业和用户立场所需的其他物品,包括其他缓冲剂,稀释剂,过滤器,针头和注射器。
在试剂盒包含他塞利昔布和第二治疗剂即化疗剂的情况下,试剂盒可以包括用于容纳分开的组合物的容器,比如分隔的瓶子或分隔的箔包装,然而,分开组合物也可以被容纳在单个未分隔的容器中。通常,试剂盒包括用于施用分开的组分的说明。当分开的组分优选以不同的剂型(例如,口服和肠胃外)以不同的给药间隔施用时,或者当处方医师期望将联合的单独组分逐步增高剂量时,试剂盒形式是特别有利的。
实施例
实施例1:p110a(α)pi3k结合测定法
结合测定法:初始偏振实验在analystht96-384(moleculardevicescorp,sunnyvale,ca.)上进行。用于荧光偏振亲和力测量的样品如下制备:将1:3连续稀释的p110αpi3k(upstatecellsignalingsolutions,charlottesville,va),由20μg/ml于偏振缓冲液(10mmtrisph7.5,50mmnacl,4mmmgcl2,0.05%chaps,和1mmdtt)中的最终浓度开始,加至10mmpip2(echelon-inc.,saltlakecity,ut.)的最终浓度。在室温温育30分钟后,通过分别加入grp-1和pip3-tamra探针(echelon-inc.,saltlakecity,ut.)至100nm和5nm的终浓度停止反应。在384孔黑色低容量
抑制剂的ic50值如下确定:将结合有pip2(10mm终浓度)的0.04mg/mlp110αpi3k(终浓度)加至孔中,所述孔中含有1:3连续稀释的拮抗剂,在偏振缓冲液中的终浓度为25mmatp(cellsignalingtechnology,inc.,danvers,ma)。在室温温育30分钟后,通过分别加入grp-1和pip3-tamra探针(echelon-inc.,saltlakecity,ut.)至100nm和5nm的终浓度停止反应。在384孔黑色低容量
或者,在放射测定法中使用浓度为1μm(微摩尔)的纯化的重组酶和atp确定对pi3k的抑制。将该化合物在100%dmso中进行连续稀释。将激酶反应物在室温温育1小时,通过加入pbs终止反应。随后使用s形剂量反应曲线拟合(可变斜率)确定ic50值。
实施例2:体外细胞增殖测定
细胞培养:在具有10%胎牛血清,100u/ml青霉素,和100μg/ml链霉素的rpmi培养基中,在标准组织培养条件下培养细胞系。hcc-1954和hdq-p1是乳腺癌细胞系(americantypeculturecollection;manassas,va)。将hcc-1954和hdq-p1细胞以800,00个细胞/孔置于6孔组织培养板的每个孔中,并在37℃温育过夜。将细胞与指定浓度的每种化合物一起温育24小时。温育后,用冷磷酸盐缓冲盐水(pbs)将细胞洗涤一次,并在补充有蛋白酶抑制剂(f.hoffmanlaroche;mannheim,germany),1mm苯甲基磺酰氟,和磷酸酶抑制剂混合物1和2(sigmaaldrich;st.louis,mo)的biosourcetm细胞提取缓冲液((invitrogen;carlsbad,ca)中裂解。使用piercebca蛋白质测定试剂盒(thermofisherscientific;rockford,il)确定蛋白质浓度。
通过使用以下方案的细胞增殖测定来测量结合pi3k的化合物的功效(mendoza等人(2002)cancerres.62:5485-5488)。
cell
该测定法评定化合物进入细胞并抑制细胞增殖的能力。该测定法的原理基于通过在均相测定法中定量存在的atp来确定活细胞数,其中加入cell
程序:第1天–接种细胞板(来自falcon#353962的带有盖子的384孔黑色,透明底部,microcleartc板),收获细胞,以1000个细胞/54μl/孔接种细胞到384孔细胞板中,进行3天测定。细胞培养基:rpmi或dmem高糖,10%胎牛血清,2mml-谷氨酰胺,p/s。在37℃,5%co2温育o/n(过夜)。
第2天-将药物添加至细胞,稀释化合物,dmso板(对于9个点以1:2连续稀释)。在96孔板的第2列中加入20μl的10mm化合物。使用来自nunc的96孔圆锥形底聚丙烯板precisionmediaplate(目录号249946),对于总共9个点在整个板中进行1:2连续稀释(10μl+20μl100%dmso)(1:50稀释)。向所有孔中加入147μl培养基。使用
向细胞加入药物,细胞板(1:10稀释):向细胞直接加入6μl的培养基+化合物(在细胞上已有54μl培养基)。在37℃,5%co2,在不经常打开的培养箱中温育3天。
第5天-培育板,在室温解冻celltiterglo缓冲液:将细胞板从37℃移出并平衡至室温,保持约30分钟。将cell
细胞活力测定法和联合测定法:将细胞以1000-2000个细胞/孔接种在384孔板中,保持16小时。在第二天,在96孔板中制备9份在dmso中的化合物的1:2连续稀释物。使用
另外的示例性体外细胞增殖测定法包括以下步骤:
1.将在培养基中含有约104个细胞(参见表3中的细胞系和肿瘤类型)的100μl细胞培养物的等分试样沉积在384孔不透明壁板的每个孔中。
2.制备含有培养基但不含细胞的对照孔。
3.将化合物加入到实验孔中,并温育3-5天。
4.将平板平衡至室温并保持约30分钟。
5.加入体积与每孔中存在的细胞培养基的体积相同的cell
6.将内容物在轨道式摇床上混合2分钟以诱导细胞裂解。
7.将板在室温下温育10分钟以稳定发光信号。
8.记录发光并以图表报告为rlu=相对发光单位。
9.使用chou和talalay联合方法以及利用
或者,将细胞以最佳密度接种在96孔板中并在测试化合物存在下温育4天。随后将alamarbluetm加入到测定培养基中,并将细胞孵育6小时,然后以544nm激发,590nm发射进行读数。使用s型剂量反应曲线拟合计算ec50值。
或者,使用cell
细胞系获自atcc(美国典型培养物保藏中心,manassas,va)或dsmz(德国微生物和细胞培养物保藏中心,braunschweig,de)。在37℃,5%co2,在补充有10%胎牛血清,100单位/ml的青霉素,2mml-谷氨酰胺和100mg/ml链霉素(lifetechnology,grandisland,ny)的rpmi1640培养基中培养细胞。
实施例3:sw48同基因细胞系活力测定
细胞培养。细胞系获自美国典型培养物保藏中心(atcc,va),或获自deutschesammlungvonmikroorganismenundzellkulturengmbh(dmsz,germany)。在37℃,5%co2,在补充有10%胎牛血清,100单位/ml的青霉素和100μg/ml链霉素的dmem或rpmi中培养细胞系。mcf7-neo/her2是在genentech开发的并且源于亲本mcf7人乳腺癌细胞系的经过体内选择的肿瘤细胞系。等基因细胞系(sw48亲本,sw48e545k和sw48h1047r)获得horizondiscoveryltd.(cambridge,uk)的许可,并在37℃,5%co2在补充有10%胎牛血清,100单位/ml的青霉素和100μg/ml链霉素的mccoy’s5a中培养。
细胞活力测定。以每孔54μl的体积以1000-2000个细胞/孔接种384孔板,然后在37℃,5%co2温育过夜(约16小时)。将化合物稀释在dmso中以产生期望的储存浓度,然后以每孔6μl的量加入。所有处理均以一式四份进行试验。在温育4天后,使用celltiter-glo(promega,madison,wi)估计活细胞的相对数,并且在wallacmultilabelreader(perkinelmer,fostercity,ca)上测量总发光。使用prism软件(graphpad,lajolla,ca)确定导致细胞活力50%抑制(ic50)或50%最大有效浓度(ec50)的药物浓度。对于未达到ic50的细胞系,列出了测试的最高浓度(10μm)。
实施例4:小鼠肿瘤异种移植体内功效
小鼠:雌性重症联合免疫缺陷小鼠(foxchase
肿瘤植入:用癌细胞启动异种移植。细胞在补充有10%胎牛血清,2mm谷氨酰胺,100单位/ml青霉素,100μg/ml硫酸链霉素和25μg/ml庆大霉素的rpmi1640培养基中培养。根据细胞系的倍增时间,在指数生长期间收获细胞,以5x106或10x106个细胞/ml的浓度重悬于磷酸盐缓冲盐水(pbs)中。将肿瘤细胞皮下植入右胁腹部,并且当平均大小接近100至150mm3的目标范围时监测肿瘤生长。在肿瘤植入后21天(指定为研究的第0天),将小鼠分成四组,每组由10只小鼠组成,所述小鼠具有75-172mm3范围的单个肿瘤体积和120-121mm3范围的组平均肿瘤体积(参见附录a)。使用以下公式计算体积:
肿瘤体积(mm3)=(w2xl)/2,其中w=肿瘤的宽度,l=肿瘤的长度(mm)。假设1mg等同于1mm3的肿瘤体积,则可估计肿瘤重量。
治疗剂:结合pi3k的化合物被提供为盐形式的干粉,其含有73%的活性剂,并在室温下避光储存。每周制备在去离子水(“溶媒”)中的0.5%甲基纤维素:0.2%吐温80中的药物剂量,并在4℃下储存。在g-033829剂量的制剂中具有含73%活性剂的盐形式。在给药的每一天,通过用无菌盐水(0.9%nacl)稀释储液的等分试样来准备结合pi3k的化合物的剂量。所有剂量均配制为递送规定的mg/kg剂量,以每20克体重0.2ml体积(10ml/kg)计算。
治疗:所有剂量均与个体动物的体重成比例,并通过每个图中所示的途径提供。
终点:使用ultracaliv卡尺(型号5410111;fredv.fowlercompany)在2维(长度和宽度)上如下测量肿瘤体积:肿瘤体积(mm3)=(长度x宽度2)x0.5,并使用excel版本11.2(microsoftcorporation)进行分析。使用线性混合效应(lme)建模方法分析来自同一动物的随着时间过去的肿瘤体积的重复测量(pinheiroj,etal.nlme:linearandnonlinearmixedeffectsmodels.rpackageversion3.192.2009;tann,etal.navitoclaxenhancestheefficacyoftaxanesinnon-smallcelllungcancermodels.clin.cancerres.2011;17(6):1394-1404)。这种方法解决了重复测量和由于在研究结束之前任何非治疗相关性动物死亡所致的少量退出。使用三次回归样条将非线性轮廓与每个剂量水平的log2肿瘤体积的时间进程拟合。然后使这些非线性轮廓与混合模型中的剂量相关。使用以下公式,计算作为溶媒对照百分比的肿瘤生长抑制(%tgi),其为相对于溶媒的每天各剂量组的拟合曲线下面积的百分比(auc):%tgi=100x(1-auc剂量/auc溶媒)。使用该公式,tgi值100%表示肿瘤停滞,tgi值>1%但<100%表示肿瘤生长延迟,tgi值>100%表示肿瘤消退。将动物的部分缓解(pr)定义为起始肿瘤体积的>50%但<100%的肿瘤消退。在研究期间的任何一天,完全缓解(cr)定义为100%肿瘤消退(即,没有可测量的肿瘤)。
毒性:在研究的前五天,每天给动物称重,之后称重为每周两次。使用adventurer
实施例5:p110α蛋白的western印迹分析
蛋白质测定法:使用piercebca蛋白质测定试剂盒(rockford,il)确定蛋白质浓度。对于免疫印迹,用nupagebistris4-12%梯度凝胶(invitrogen;carlsbad,ca)通过电泳分离相等的蛋白质量;使用来自invitrogen的iblot系统和方案将蛋白质转移到硝酸纤维素膜上。从cellsignaling(danvers,ma)获得p110α和phospho-akt(ser473)的抗体。β肌动蛋白和gapdh的抗体来自sigma。
对于western印迹,用nupagetris乙酸盐的3-18%梯度凝胶(invitrogen)进行电泳来分离等量的蛋白质。使用来自invitrogen(carlsbad,ca)的iblot系统和方案将蛋白质转移到硝酸纤维素孔膜上。pakt(ser473),p110α和p85抗体获自cellsignalingtechnology(danvers,ma)。β肌动蛋白抗体获自sigma-aldrich(st.louis,mo)。使用增强的化学发光western印迹检测试剂(gehealthcarelifesciences,pittsburgh,pa),用辣根过氧化物酶缀合的二级抗体igg检测特异性抗原-抗体相互作用。
遵循制造商的方案(pi3激酶p110α抗体#4255,pi3激酶p110α(c73f8)兔mab#4249,cellsignalingtechnology),使用多克隆兔抗pi3kp110α抗体,进行western印迹分析。单克隆和多克隆pi3kp110α抗体是可商购的(santacruzbiotechnology)。参见popkie等人(2010)jbiolchem.;285(53):41337-47;yoshioka等人(2012)natmed.oct;18(10):1560-9;biswas等人(2013)jbiolchem.jan25;288(4):2325-39;ramadani等人(2010)scisignal.aug10;3(134)。
western印迹方案(细胞信号传导技术):
对于western印迹,将所述膜与5%w/vbsa,1xtbs,0.1%
稀释度:
western印迹,1:1000,
免疫沉淀,1:50
免疫组织化学,1:400
提供在10mmhepes钠(ph7.5),150mmnacl,100μg/mlbsa,50%甘油和小于0.02%的叠氮化钠中。储存于-20℃。不要分装抗体。
a.溶液和试剂
注:用反渗透去离子(rodi)或相当级别的水制备溶液。
1.20x磷酸盐缓冲盐水(pbs):(#9808)为了制备1l1xpbs:将50ml20xpbs加入到950mldh2o中,混合。
2.10xtris缓冲盐水(tbs):(#12498)为了制备1l1xtbs:将100ml10xtbs加入到900mldh2o中,混合。
3.1xsds样品缓冲液:blueloadingpack(#7722)或redloadingpack(#7723);通过将1/10体积的30xdtt加入到1体积的3xsds上样缓冲液中来制备新鲜的3x还原上样缓冲液。用dh2o稀释至1x。
4.10xtris甘氨酸sds运行缓冲液:(#4050)为了制备1l1x运行缓冲液:将100ml10x运行缓冲液加入到900mldh2o中,混合。
5.10xtris-甘氨酸转移缓冲液:(#12539)制备1l1x转移缓冲液:将100ml10x转移缓冲液加入到200ml甲醇+700mldh2o中,混合。
6.具有
7.脱脂奶粉:(#9999)。
8.封闭缓冲液:1xtbst,含5%w/v脱脂奶粉;对于150ml,将7.5g脱脂奶粉加入到150ml1xtbst中并充分混合。
9.洗涤缓冲液:(#9997)1xtbst。
10.牛血清白蛋白(bsa):(#9998)。
11.一级抗体稀释缓冲液:含5%bsa的1xtbst;对于20ml,将1.0gbsa加入到20ml1xtbst中,并充分混合。
12.生物素化蛋白质梯子检测包装:(#7727)。
13.预染色蛋白质标记,宽范围(预混合格式):(#7720)。
14.印迹膜和纸:(cellsignalingtechnology#12369)本方案已针对硝酸纤维素膜进行了优化。通常推荐0.2μm孔径。
15.与hrp缀合的二级抗体:抗兔igg,hrp连接的抗体(#7074)。
16.检测试剂:signalfiretmecl试剂(#6883)。
b.蛋白质印迹
样品制备的一般方案。
1.通过加入含有调节剂的新鲜培养基来处理细胞,持续期望的时间。
2.从培养物吸出培养基;用1xpbs洗涤细胞;吸出。
3.通过加入1xsds样品缓冲液(6孔板每孔100μl,或直径为10cm的平板500μl)来裂解细胞。立即从板上刮下细胞并将提取物转移到微量离心管中。保持在冰上。
4.超声处理10-15秒以完成细胞裂解和剪切dna(以降低样品粘度)。
5.将20μl样品在95-100℃下加热5分钟;放在冰上冷却。
6.微量离心机内离心5分钟。
7.将20μl上样到sds-page凝胶(10cmx10cm)上。
注:推荐将预染色分子量标记(#7720,10μl/泳道)上样以验证电转移,并将生物素化蛋白质梯(#7727,10μl/泳道)上样以确定分子量。
8.电转移到硝酸纤维素膜(#12369)。
c.膜封闭和抗体温育
注:膜的体积为10cmx10cm(100cm2);对于不同尺寸的膜,相应地调整体积。
i.膜封闭
1.(任选)转移后,在室温下用25mltbs洗涤硝酸纤维素膜5分钟。
2.在室温下将膜在25ml封闭缓冲液中温育1小时。
3.用15mltbst洗涤3次,每次5分钟。
ii.一级抗体温育
1.将膜和一级抗体(按照产品数据表中推荐的适当稀释度和稀释剂)在10ml一级抗体稀释缓冲液中在4℃温和搅拌下温育过夜。
2.用15mltbst洗涤3次,每次5分钟。
3.在10ml封闭缓冲液中,用抗兔igg,hrp连接的抗体(#7074,以1:2000)和抗生物素,hrp连接的抗体(#7075,以1:1000–1:3000)温育膜,在室温下温和搅拌1小时,以检测生物素化蛋白质标记物。
4.用15mltbst洗涤3次,每次5分钟。
5.继续进行检测(d节)。
d.蛋白质检测
使用说明:
1.在tbst中洗涤膜结合的hrp(抗体缀合物)三次,持续5分钟。
2.通过稀释一份2x试剂a和一份2x试剂b(例如,对于10ml,加入5ml试剂a和5ml试剂b)来制备1xsignalfiretmecl试剂(#6883)。充分混合。
3.将底物与膜温育1分钟,除去过量溶液(膜保持湿润),包裹在塑料中并暴露于x射线胶片。
*避免反复接触皮肤。
western印迹再探测方案
现有膜的再探测是当只有有限量的样品可用时独立地进行多种蛋白质的免疫印迹的便利手段。需要指出的是,为了获得最佳可能的结果,始终推荐新的印迹。再探测可以是一种有价值的方法,但是印迹的每次再探测可能会增加背景信号。另外,建议您在再探测之前验证第一抗体复合物的去除,以便使归因于新抗体结合的信号不是来自第一次免疫印迹实验的残留信号。这可以通过如下方式来完成:使印迹再暴露于ecl试剂并确保在添加后续一级抗体之前没有信号。
a.溶液和试剂
注:用反渗透去离子(rodi)或等效纯化的水制备溶液。
1.洗涤缓冲液:具有
2.剥离缓冲液:为了制备100ml,将0.76gtris碱,2gsds和700μlβ-巯基乙醇混合。用去离子h2o加至100ml。用hcl将ph调节至6.8。
b.方案
1.在胶片曝光后,在tbst中洗涤膜四次,每次5分钟。如果不允许膜干燥,则获得最佳结果。
2.在50℃下在剥离缓冲液(伴有轻微搅拌)中温育膜30分钟。
3.在tbst中洗涤膜6次,每次5分钟。
4.(任选)为了确保原始信号被去除,用10mltbst洗涤膜两次,每次5分钟。用
5.在tbst中再洗涤膜4次,每次5分钟。
6.膜现在可以再使用了。在western免疫印迹方案中的“膜封闭和抗体温育”步骤开始检测。
实施例6:突变型p110α的免疫组织化学(ihc)检测
为了确定患者肿瘤活检样品中的突变型p110α水平,可以进行各种诊断测定。在一个实施方案中,可以通过免疫组织化学(ihc)分析突变型p110α水平。来自肿瘤活检的石蜡包埋的组织切片可以接受ihc测定,并符合如下染色强度标准:
得分0-观察不到染色或在少于10%的肿瘤细胞中观察到膜染色。
得分1+-在超过10%的肿瘤细胞中检测到微弱/几乎不可察觉的膜染色。仅在膜的一部分中细胞被染色。
得分2+-在超过10%的肿瘤细胞中观察到弱至中等的完全膜染色。
得分3+-在超过10%的肿瘤细胞中观察到中等至强的完全膜染色。
肿瘤样品可根据其得分加以表征。
在一些实施方案中,使用抗突变型p110α抗体检测突变型p110α生物标志物。在一些实施方案中,通过ihc检测突变型p110α生物标志物为弱染色强度。在一些实施方案中,通过ihc检测突变型p110α生物标志物为中等染色强度。在一些实施方案中,通过ihc检测突变型p110α生物标志物为强染色强度。
可以使用ventana
实施例7:p110α细胞系的质谱分析
对于来自用dmso(溶媒)或gdc-0032(500nm)各自处理24小时的三个细胞系的免疫沉淀的p110α(pik3ca)蛋白,进行了液相色谱-串联质谱(lc-ms/ms)分析,所述三个细胞系为:hcc1954,hcc202和hdqp1。对于n=4的生物学重复,以每细胞系/处理(总共6个样品/实验)4-6mg蛋白裂解物开始进行每一实验。基于相邻泳道中纯化的p110αl蛋白的迁移,切下每样品对应于pik3ca的预期迁移的一个凝胶区域。将凝胶块切成约1mm3的方丁,并如下进行凝胶内消化。将凝胶块用50mm碳酸氢铵/50%乙腈脱去染色并用100%乙腈脱水,然后使用50mm二硫苏糖醇(30分钟,50℃)和50mm碘乙酰胺(20分钟,室温)分别还原和烷基化。再次将凝胶块脱水,然后使其在50mm碳酸氢铵/5%乙腈消化缓冲液中的20ng/μl胰蛋白酶中在冰上再溶胀2小时,然后转移到37℃的烘箱中温育过夜。将消化的肽转移到微量离心管中,将凝胶块提取两次:用50%乙腈/0.5%三氟乙酸提取一次,再用100%乙腈进行第二轮提取。将提取物与消化的肽合并,真空离心(speed-vac)干燥至完成。在lc-ms/ms分析前30分钟,将样品在5%甲酸/0.1%七氟丁酸/0.01%过氧化氢中复原。
在与watersnanoacquityuplc偶联的thermoltqorbitrapelite上,通过利用重复注射(除第一次重复注射一次样品外)的lc-ms/ms分析样品。将肽上样到装有1.7umbeh-130树脂的0.1mm×100mmwaterssymmetryc18柱上,并通过两步线性梯度分离,其中溶剂b(98%乙腈,2%水)经过20分钟从5%缓慢上升至25%,然后经过2分钟从25%上升至50%。以数据依赖性模式获取数据,其中以60,000分辨率收集的orbitrap全ms扫描和选择用于在离子阱中的cidms/ms碎裂的前15个最强的前体。使用mascot搜索ms2光谱,两者都针对人类蛋白质的连接的目标-诱饵uniprot数据库,以及包含野生型e545k和h1047r突变型pik3ca序列的小型数据库,以便鉴定突变型肽。在人工检验之前,使用线性判别分析,以10%的容许错误发现率过滤uniprot搜索的光谱匹配。
使用内部软件(msplorer),以10ppm质量公差生成pik3ca肽的提取离子色谱图和峰面积积分。在每个块的基础上,将14个肽中每一者的峰面积数据标准化为六个样品中最丰富的峰面积。在重复注射(技术重复)可用的情况下,将两个重复的标准化数据平均以产生每个肽条件实验(即定量特征)的单个标准化峰面积,以生成蛋白质序列图。
对于统计学分析,使用线性混合效应建模(lme4包)将四个生物学重复中的原始非标准化峰面积统一为r,以确定对于总pik3ca,野生型pik3ca和突变型pik3ca每一者按照细胞系比较dmso(溶媒)与gdc-0032(500nm)处理的相对比率和p值。基于从以下肽生成的数据确定总pik3ca:
对于携带h1047r突变的细胞系(即hcc-1954),基于qmndahhggwttk(1042-1054;749.82824m/z)肽(seqidno.:7)确定了野生型pik3ca,并且基于覆盖1047基因座的qmndar(1042-1047;375.66360m/z)(seqidno.:18)和hggwttk(1048-1054;393.6983m/z)(seqidno.:8)肽确定了突变型pik3ca(图24a和24b)。对于携带e545k突变的细胞系(即hcc-202),基于dplseiteqek(538-548;644.81917m/z)(seqidno.:19)肽确定了野生型pik3ca,并且基于覆盖545基因座的dplseitk(538-545;451.74627m/z)(seqidno.:20)肽确定了突变型pik3ca。当确定总pik3ca时,不考虑这两个基因座的肽。在适用的情况下,将pik3ca肽中半胱氨酸残基以其脲基甲基化形式(+57.021da)进行分析,并且将甲硫氨酸残基根据其单氧化(met-亚砜)形式(+15.9949da)进行定量。使用总pik3ca,野生型pik3ca和突变型pik3ca的log2比率(gdc-0032/dmso)和相应p值,通过应用质量守恒原理来确定每一条件和细胞系的野生型和突变型pik3ca的相对丰度(和相关的95%置信区间)。明确地:
wt_pi3kca_dmso+mut_pi3kca_dmso=总_pi3kca_dmso
wt_pi3kca_gdc+mut_pi3kca_gdc=总_pi3kca_gdc
wt_pi3kca比例=p=wt_pi3kca/总_pi3kca
mut_pi3kca比例=1-p=mut_pi3kca/总_pi3kca
将数据绘制为相对强度值,其中总_pi3kca_dmso被标准化为1.0,并且误差条代表基于线性效应模型的每个测量值的95%置信区间。
虽然为了清楚理解的目的,通过说明和实施例的方式详细描述了前述发明,但是说明书和实施例不应被解释为限制本发明的范围。本文中引用的所有专利和科学文献的公开内容通过提述以其整体明确地并入本文。
序列表
<110>k·埃德加(edgar,kyle)
l·弗里德曼(friedman,lori)
d·桑派斯(sampath,deepak)
k·宋(song,kyung)
i·沃茨(wertz,ingrid)
t·威尔逊(wilson,timothy)
<120>用他塞利昔布进行治疗的方法
<130>p32910-us-1
<141>2016-06-28
<150>us62/186,236
<151>2015-06-29
<160>20
<210>1
<211>17
<212>dna
<213>人工序列
<220>
<223>序列是合成的
<400>1
atgatgcacatcatggt17
<210>2
<211>24
<212>dna
<213>人工序列
<220>
<223>序列是合成的
<400>2
ggctttggagtatttcatgaaaca24
<210>3
<211>25
<212>dna
<213>人工序列
<220>
<223>序列是合成的
<400>3
gaagatccaatccatttttgttgtc25
<210>4
<211>16
<212>dna
<213>人工序列
<220>
<223>序列是合成的
<400>4
tgatgcacgtcatggt16
<210>5
<211>24
<212>dna
<213>人工序列
<220>
<223>序列是合成的
<400>5
ggctttggagtatttcatgaaaca24
<210>6
<211>25
<212>dna
<213>人工序列
<220>
<223>序列是合成的
<400>6
gaagatccaatccatttttgttgtc25
<210>7
<211>13
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>7
glnmetasnaspalahishisglyglytrpthrthrlys
510
<210>8
<211>7
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>8
hisglyglytrpthrthrlys
5
<210>9
<211>8
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>9
glualathrleuilethrilelys
5
<210>10
<211>8
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>10
aspleuasnserprohisserarg
5
<210>11
<211>9
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>11
leucysvalleuglutyrglnglylys
5
<210>12
<211>11
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>12
valcysglycysaspglutyrpheleuglulys
510
<210>13
<211>7
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>13
valprocysserasnproarg
5
<210>14
<211>14
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>14
glualaglyphesertyrserhisalaglyleuserasnarg
510
<210>15
<211>11
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>15
tyrgluglntyrleuaspasnleuleuvalarg
510
<210>16
<211>10
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>16
pheglyleuleuleuglusertyrcysarg
510
<210>17
<211>9
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>17
leuileasnleuthraspileleulys
5
<210>18
<211>6
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>18
glnmetasnaspalaarg
5
<210>19
<211>11
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>19
aspproleusergluilethrgluglnglulys
510
<210>20
<211>8
<212>prt
<213>人工序列
<220>
<223>序列是合成的
<400>20
aspproleusergluilethrlys
5
- 还没有人留言评论。精彩留言会获得点赞!