龙眼叶降血糖有效部位活性成分的提取分离方法与流程
本发明涉及植物有效成分的分离技术领域,具体涉及龙眼叶降血糖有效部位活性成分的提取分离方法。
背景技术:
龙眼叶为龙眼属龙眼(dimocarpuslonganlour.)的叶或嫩芽。味甘、淡,性平,具有发表清热、利湿解毒之功效,主治感冒发热、疟疾、疔疮、湿疹等。龙眼原产于我国南部和越南北部的南亚热带区域,在我国已有2000多年栽培历史。我国龙眼栽培面积和产量均居世界首位。每年龙眼被摘之后,果农对树木进行截枝,大量的树叶被抛弃或燃烧,造成资源浪废和环境污染。龙眼叶主要含有鞣质、酚酸类、黄酮类及挥发油等成分,发明人曾对龙眼叶进行了系统的化学成分研究,发现其主要活性成分为黄酮成分。现代药理研究表明龙眼叶具有很好的降血糖作用,我国南方龙眼叶资源蕴藏量丰富,但国内外关于龙眼叶降糖作用的研究报道较少,发明人曾对其降糖作用进行过研究,采用体外抑制α-葡萄糖苷酶活性试验,发现羽扇豆醇、没食子酸乙酯、山柰酚、木犀草素、槲皮素等成分对α-葡萄糖苷酶均有较强有抑制作用。目前国内对龙眼叶的研究报道不多,主要集中在黄酮类成分的提取方面,未见针对其降血糖有效部位或活性成分进行提取分离方法。
以上背景技术内容的公开仅用于辅助理解本发明的发明构思及技术方案,其并不必然属于本专利申请的现有技术,在没有明确的证据表明上述内容在本专利申请的申请日已经公开的情况下,上述背景技术不应当用于评价本申请的新颖性和创造性。
技术实现要素:
本发明的目的在于提供一种从龙眼叶中提取分离具有降血糖活性成分的方法。
为实现上述目的,本发明所采用的技术方案如下:
一种龙眼叶降血糖有效部位活性成分,该活性成分由下述方法制备而得:龙眼叶经粉碎后用乙醇渗漉提取,渗漉液减压回收乙醇后得浸膏,加水将浸膏混悬,依次用石油醚、乙酸乙酯萃取得到相应的石油醚部位、乙酸乙酯部位,取石油醚部位、乙酸乙酯部位通过反复硅胶柱色谱分离纯化,重结晶,即得。
进一步的,以上所述的龙眼叶降血糖有效部位活性成分,包括羽扇豆醇、没食子酸乙酯、山柰酚、木犀草素、槲皮素。
进一步的,以上所述的龙眼叶降血糖有效部位活性成分,具有抑制α-葡萄糖苷酶的作用。
一种以上所述的龙眼叶降血糖有效部位活性成分的制备方法,包括以下步骤:
s1.取干燥龙眼叶,粉碎后用乙醇渗漉提取,渗漉提取液减压回收乙醇,得浸膏;
s2.将乙醇浸膏加水混悬,依次用石油醚、乙酸乙酯各萃取3次,得相应萃取液;
s3.分取各萃取液减压浓缩回收溶剂,分别得到石油醚部位、乙酸乙酯部位;
s4.取石油醚部位,经硅胶柱色谱,以石油醚-乙酸乙酯(100:0→0:100)梯度洗脱,经tlc检识,不同展开剂展开,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到馏分fr.1~15共15个馏分;
s5.取馏分fr.5,重结晶得到化合物1羽扇豆醇;
s6.取乙酸乙酯部位,经硅胶柱色谱,依次用氯仿-乙酸乙酯(50:1→7:1)、氯仿-甲醇(5:1)进行梯度洗脱,将所得洗脱液经tlc检识,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到fra.1~fra.8共8个馏分;
s7.取馏分fra.4,重结晶后得到化合物2没食子酸乙酯、化合物3山柰酚;
s8.取馏分fra.5,经过硅胶柱色谱,以氯仿-乙酸乙酯(50:1→1:1)梯度洗脱,得到fra.5.1~5.6共6个馏分;
s9.取馏分fra.5.6,重结晶后,得化合物2没食子酸乙酯、化合物4木犀草素;
s10.取馏分fra.5.5,经硅胶柱色谱,氯仿:甲醇=70:1-50:1-30:1-10:1-5:1-1:1-甲醇梯度洗脱,重结晶后,得到化合物2没食子酸乙酯、化合物5槲皮素。
进一步的,以上所述的龙眼叶降血糖有效部位活性成分的制备方法,所述乙醇为50%乙醇-无水乙醇。
更进一步的,以上所述的龙眼叶降血糖有效部位活性成分的制备方法,所述乙醇渗漉提取为依次用95%乙醇和50%乙醇渗漉提取。
本发明的有益效果在于:
1、本发明所用的原材料为龙眼的叶或嫩芽,现有技术中大量的龙眼叶被废弃或燃烧,造成资源浪费和环境污染,本发明变废为宝,原材料来源丰富,价格低廉。
2、本发明提供的具有降血糖作用的龙眼叶有效部位活性成分的提取分离方法,工艺设计合理、提取有效成分含量高,可操作性强,适合工业化大生产。
附图说明
图1为pnp标准工作曲线。
图2为实施例1制备所得化合物对α-葡萄糖苷酶抑制率比较图。
图3为实施例1制备所得化合物不同浓度对α-葡萄糖苷酶抑制活性的影响图。
下面结合具体实施例对本发明作进一步描述,但不限制本发明的保护范围和应用范围:一、龙眼叶降血糖有效部位活性成分的制备
实施例1
龙眼叶降血糖有效部位活性成分的制备方法,包括以下步骤:
s1.取干燥龙眼叶10kg,粉碎后依次用95%乙醇和50%乙醇渗漉提取,渗漉提取液减压回收乙醇得浸膏;
s2.将乙醇浸膏加水混悬,依次用石油醚、乙酸乙酯各萃取3次,得相应提取液;
s3.分取各提取液减压浓缩回收溶剂,分别得到石油醚部位、乙酸乙酯部位;
s4.取石油醚部位,经硅胶柱色谱,以石油醚-乙酸乙酯(100:0→0:100)梯度洗脱,经tlc检识,不同展开剂展开,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到馏分fr.1~15共15个馏分;
s5.取馏分fr.5(30:1)中得到白色长针状、白色针状、白色粉末,重结晶得到化合物1羽扇豆醇;
s6.取乙酸乙酯部位,经硅胶柱色谱,依次用氯仿-乙酸乙酯(50:1→7:1)、氯仿-甲醇(5:1)进行梯度洗脱,将所得洗脱液经tlc检识,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到fra.1~fra.8共8个馏分;
s7.取馏分fra.4(氯仿:乙酸乙酯=7:1)中得到白色长针状晶体,重结晶后得到化合物2没食子酸乙酯、化合物3山柰酚;
s8.取馏分fra.5(氯仿-甲醇=5:1)用硅胶柱色谱分离,以氯仿-乙酸乙酯(50:1→1:1)梯度洗脱,得到fra.5.1~5.6共6个馏分;
s9.取馏分fra.5.6,经硅胶柱色谱分离,以(氯仿:甲醇=30:1→甲醇)梯度洗脱,得白色粉末,重结晶后,得化合物2没食子酸乙酯、化合物4木犀草素;
s10.取馏分fra.5.5,经硅胶柱色谱分离,以(氯仿:甲醇=70:1-50:1-30:1-10:1-5:1-1:1-甲醇)梯度洗脱,得白色针状结晶、白色粉末,重结晶后,分别从(氯仿:甲醇=30:1)得到化合物2没食子酸乙酯、(氯仿:甲醇=10:1)得到化合物5槲皮素。
实施例2
龙眼叶降血糖有效部位活性成分的制备方法,包括以下步骤:
s1.取干燥龙眼叶10kg,粉碎后用50%乙醇渗漉提取,合并渗漉提取液,减压回收乙醇得浸膏;
s2.将乙醇浸膏加水混悬,依次用石油醚、乙酸乙酯各萃取3次,得相应提取液;
s3.分取各提取液减压浓缩回收溶剂,分别得到石油醚部位、乙酸乙酯部位;
s4.取石油醚部位,经硅胶柱色谱,以石油醚-乙酸乙酯(100:0→0:100)梯度洗脱,经tlc检识,不同展开剂展开,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到馏分fr.1~15共15个馏分;
s5.取馏分fr.5(30:1)中得到白色长针状、白色针状、白色粉末,重结晶得到化合物1羽扇豆醇;
s6.取乙酸乙酯部位,经硅胶柱色谱,依次用氯仿-乙酸乙酯(50:1→7:1)、氯仿-甲醇(5:1)进行梯度洗脱,将所得洗脱液经tlc检识,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到fra.1~fra.8共8个馏分;
s7.取馏分fra.4(氯仿:乙酸乙酯=7:1)中得到白色长针状晶体,重结晶后得到化合物2没食子酸乙酯、化合物3山柰酚;
s8.取馏分fra.5(氯仿-甲醇=5:1)用硅胶柱色谱分离,以氯仿-乙酸乙酯(50:1→1:1)梯度洗脱,得到fra.5.1~5.6共6个馏分;
s9.取馏分fra.5.6,经硅胶柱色谱分离,以(氯仿:甲醇=30:1→甲醇)梯度洗脱,得白色粉末,重结晶后,得化合物2没食子酸乙酯、化合物4木犀草素;
s10.取馏分fra.5.5,经硅胶柱色谱分离,以(氯仿:甲醇=70:1-50:1-30:1-10:1-5:1-1:1-甲醇)梯度洗脱,得白色针状结晶、白色粉末,重结晶后,从(氯仿:甲醇=30:1)得到化合物2没食子酸乙酯、(氯仿:甲醇=10:1)得到化合物5槲皮素。
实施例3
龙眼叶降血糖有效部位活性成分的制备方法,包括以下步骤:
s1.取干燥龙眼叶10kg,粉碎后用无水乙醇渗漉提取,渗漉提取液减压回收乙醇得浸膏;
s2.将乙醇浸膏加水混悬,依次用石油醚、乙酸乙酯各萃取3次,得相应提取液;
s3.分取各提取液减压浓缩回收溶剂,分别得到石油醚部位、乙酸乙酯部位;
s4.取石油醚部位,经硅胶柱色谱,以石油醚-乙酸乙酯(100:0→0:100)梯度洗脱,经tlc检识,不同展开剂展开,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到馏分fr.1~15共15个馏分;
s5.取馏分fr.5(30:1)中得到白色长针状、白色针状、白色粉末,重结晶得到化合物1羽扇豆醇;
s6.取乙酸乙酯部位,经硅胶柱色谱,依次用氯仿-乙酸乙酯(50:1→7:1)、氯仿-甲醇(5:1)进行梯度洗脱,将所得洗脱液经tlc检识,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到fra.1~fra.8共8个馏分;
s7.取馏分fra.4(氯仿:乙酸乙酯=7:1)中得到白色长针状晶体,重结晶后得到化合物2没食子酸乙酯、化合物3山柰酚;
s8.取馏分fra.5(氯仿-甲醇=5:1)用硅胶柱色谱分离,以氯仿-乙酸乙酯(50:1→1:1)梯度洗脱,得到fra.5.1~5.6共6个馏分;
s9.取馏分fra.5.6,经硅胶柱色谱分离,以(氯仿:甲醇=30:1→甲醇)梯度洗脱,得白色粉末,重结晶后,得化合物2没食子酸乙酯、化合物4木犀草素;
s10.取馏分fra.5.5,经硅胶柱色谱分离,以(氯仿:甲醇=70:1-50:1-30:1-10:1-5:1-1:1-甲醇)梯度洗脱,得白色针状结晶、白色粉末,重结晶后,从(氯仿:甲醇=30:1)得到化合物2没食子酸乙酯、(氯仿:甲醇=10:1)得到化合物5槲皮素。
实施例4
龙眼叶降血糖有效部位活性成分的制备方法,包括以下步骤:
s1.取干燥龙眼叶10kg,粉碎后用75%乙醇渗漉提取,渗漉提取液减压回收乙醇得浸膏;
s2.将乙醇浸膏加水混悬,依次用石油醚、乙酸乙酯各萃取3次,得相应提取液;
s3.分取各提取液减压浓缩回收溶剂,分别得到石油醚部位、乙酸乙酯部位;
s4.取石油醚部位,经硅胶柱色谱,以石油醚-乙酸乙酯(100:0→0:100)梯度洗脱,经tlc检识,不同展开剂展开,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到馏分fr.1~15共15个馏分;
s5.取馏分fr.5(30:1)中得到白色长针状、白色针状、白色粉末,重结晶得到化合物1羽扇豆醇;
s6.取乙酸乙酯部位,经硅胶柱色谱,依次用氯仿-乙酸乙酯(50:1→7:1)、氯仿-甲醇(5:1)进行梯度洗脱,将所得洗脱液经tlc检识,碘蒸气显色,将相同斑点和rf值相同的馏分合并,得到fra.1~fra.8共8个馏分;
s7.取馏分fra.4(氯仿:乙酸乙酯=7:1)中得到白色长针状晶体,重结晶后得到化合物2没食子酸乙酯、化合物3山柰酚;
s8.取馏分fra.5(氯仿-甲醇=5:1)用硅胶柱色谱分离,以氯仿-乙酸乙酯(50:1→1:1)梯度洗脱,得到fra.5.1~5.6共6个馏分;
s9.取馏分fra.5.6,经硅胶柱色谱分离,以(氯仿:甲醇=30:1→甲醇)梯度洗脱,得白色粉末,重结晶后,得化合物2没食子酸乙酯、化合物4木犀草素;
s10.取馏分fra.5.5,经硅胶柱色谱分离,以(氯仿:甲醇=70:1-50:1-30:1-10:1-5:1-1:1-甲醇)梯度洗脱,得白色针状结晶、白色粉末,重结晶后,从(氯仿:甲醇=30:1)得到化合物2没食子酸乙酯、(氯仿:甲醇=10:1)得到化合物5槲皮素。
二、实施例1制备所得化合物的结构鉴定
化合物1:白色针状结晶。与羽扇豆醇对照品共薄层,三个不同的展开系统展开,其rf值及显色行为一致,故鉴定化合物1为羽扇豆醇。
化合物2:白色粉末。ei-msm/z:198[m]+,分子式c9h10o5。1h-nmr(500mhz,pyr-d6)δ:7.19(2h,s,h-2),7.19(2h,s,h-6),4.24(2h,q,j=7.1hz,h-8),1.15(3h,t,j=7.1hz,h-9);13c-nmr(500mhz,pyr-d6)δ:121.5(c-1),110.2(c-2),147.7(c-3),140.9(c-4),147.7(c-5),110.2(c-6),167.2(c-7),60.5(c-8),14.5(c-9)。以上数据与文献报道对照基本一致,故鉴定化合物2为没食子酸乙酯。
化合物3:黄色粉末。ei-msm/z:286[m]+,分子式c15h10o6。与山柰酚对照品共薄层,三个不同的展开系统展开,其rf值及显色行为一致,故鉴定化合物3为山柰酚。
化合物4:黄色针状结晶,ei-msm/z:286[m]+,分子式c15h10o6。与木犀草素对照品共薄层,三个不同的展开系统展开,其rf值及显色行为一致,故鉴定化合物4为木犀草素。
化合物5:黄色粉末。ei-msm/z:302[m]+,分子式c15h10o7。1h-nmr(500mhz,meod)δ:6.17(1h,d,j=2.1hz,h-6),6.38(1h,d,j=2.1hz,h-8),7.73(1h,d,j=2.2hz,h-2'),6.87(1h,d,j=8.5hz,h-5'),7.62(1h,dd,j=8.5、2.15hz,h-6');13c-nmr(500mhz,meod)δ:147.9(c-2),137.2(c-3),177.3(c-4),162.5(c-5),99.2(c-6),165.6(c-7),94.4(c-8),158.2(c-9),104.5(c-10),121.6(c-1'),115.9(c-2'),146.2(c-3'),148.8(c-4'),116.2(c-5'),124.1(c-6')。以上数据与文献报道对照基本一致,故鉴定化合物5为槲皮素。
三、实施例1制备所得化合物对对α-葡萄糖苷酶抑制作用的研究
1仪器与材料
1.1仪器
infinitem200pro酶标仪;lrh-250-s恒温恒湿培养箱(韶关市泰宏医疗器械有限公司);kq-500da超声仪(昆山市超声仪器有限公司);hws-24电热恒温水浴锅(上海齐欣科学仪器有限公司);bsa224s电子天平
1.2材料
试药:α-葡萄糖苷酶(g5003-100unsigma公司,批号slbs9070);对硝基苯基-α-d-吡喃葡萄糖苷(pnpg,sigma公司,批号bcbr9743v);对硝基苯酚(pnp,天津市大茂化学试剂厂,批号:20170301);还原型谷胱甘肽(gsh,sigma公司,批号213e053);阿卡波糖(acarbose,拜耳公司,批号bj33686);96微孔板;各种移液器及枪头;二甲亚砜(dmso)、无水碳酸钠、磷酸二氢钾、磷酸氢二钾均为分析纯。
样品:取实施例1分离纯化所得的6个化合物作为样品。
2方法
2.1供试品溶液制备
分别精密称取适量的实施例1制备所得化合物:羽扇豆醇、没食子酸乙酯、山柰酚、木犀草素、槲皮素,用dmso溶解,使得浓度为9.9mg/ml、10.1mg/ml、10.0mg/ml、10.2mg/ml、9.6mg/ml。
2.2反应溶液的配制
0.1mmol/l磷酸盐缓冲溶液(pbs,ph=6.8)、0.57u/mlα-葡萄糖苷酶、1mg/mlgsh、20mmol/lpnpg、1000μmol/lpnp、0.2mol/lnaco3、10mg/ml阿卡波糖为阳性对照。
2.3α-葡萄糖苷酶活性测定
方法:微孔板法
将2.1、2.2中配制好的样品和反应液,在96微孔板上进行,反应体系如下:取ph6.8的磷酸钾缓冲液50μl,分别加入gsh10μl,样品10μl,0.57u/mlα-葡萄糖苷酶5μl,37℃恒温反应15min,加入浓度为20mmol/lpnpg20μl,37℃恒温反应15min,再加入浓度为0.2mol/l的naco3溶液40μl,于400nm波长下测定od值,每个样品平行测定5次。以阿卡波糖为阳性对照,按下式计算抑制率:
酶活性抑制率(%)=[a空白-(a样品-a背景)]/a空白×100
注:a空白:无样品加入反应的吸光值;a样品:加入样品反应的吸光值;a背景:无酶液加入的吸光值。计算抑制率,并用spss22.0软件求出相应ic50值。
2.4标准曲线的绘制
根据采用的反应体系,用磷酸钾缓冲液(ph6.8)配置1000μmol/lpnp,分别稀释成400μmol/l、300μmol/l、200μmol/l、100μmol/l、80μmol/l、40μmol/l、20μmol/l、0μmol/l。分别取9种不同浓度的pnp溶液各160μl,加入0.2mol/lnaco3溶液80μl,混匀,在400nm波长下测定od值,测3组取平均值。以od值为纵坐标,pnp浓度为横坐标,做标准曲线。
3结果
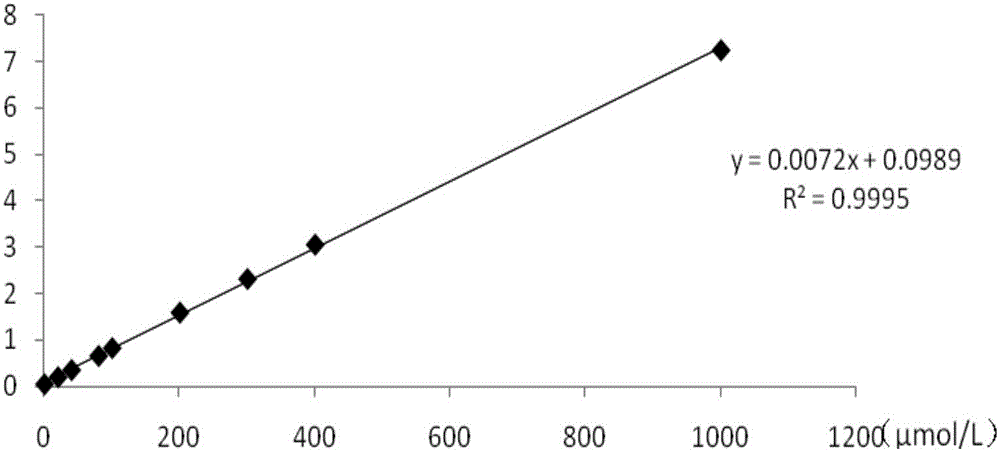
3.1pnp标准工作曲线的绘制
结果见附图1。
图1显示求得pnp标准工作曲线的回归方程为y=0.0072x+0.0989,r2=0.9995,说明用pnp在0μmol/l-1000μmol/l与od值呈现良好的线性关系。
3.2实施例1制备所得化合物对α-葡萄糖苷酶抑制作用比较
结果见表1及附图2。
表1实施例1制备所得化合物对α-葡萄糖苷酶抑制活性(n=5)
由表1可知,以阿卡波糖为阳性对照,α-葡萄糖苷酶抑制活性结果显示,实施例1制备所得化合物:羽扇豆醇、没食子酸乙酯、山柰酚、木犀草素、槲皮素抑制率分别达到65.88%(ic50=60.75mg/l)、71.64%(ic50=49.62mg/l)、65.86%(ic50=56.08mg/l)、72.50%(ic50=27.87mg/l)、67.99%(ic50=56.48mg/l),均比阳性对照药阿卡波糖63.02%(ic50=70.51mg/l)高,由此说明本发明实施例1制备所得化合物对α-葡萄糖苷酶抑制活性均较高,而且这些化合物主要分布在乙酸乙酯部位。
图2显示,在同一浓度下(0.5mg/ml),实施例1制备所得化合物:羽扇豆醇、没食子酸乙酯、山柰酚、木犀草素、槲皮素对α-葡萄糖苷酶抑制率均高于阿卡波糖(如表1)。
3.3实施例1制备所得化合物不同浓度对α-葡萄糖苷酶抑制活性的影响
结果见附图3。
图3显示,实施例1制备所得化合物对α-葡萄糖苷酶抑制活性随浓度的增加而增加,其中没食子酸乙酯、山柰酚、木犀草素、槲皮素浓度在0.1-1mg/ml大体上是呈直线增加,当浓度增加到一定时,浓度只有很小变化。在实验浓度范围内,阿卡波糖的抑制率也随浓度的增加而增加。
- 还没有人留言评论。精彩留言会获得点赞!