活性药物成分的连续络合的制作方法
本发明概况地涉及活性药物成分(api)的络合物,特别涉及基于一种或多种环糊精的那些络合物,以及涉及提供这种络合物的改进方法。
背景技术
几种活性药物成分具有低水溶性,这降低了它们的生物利用度和在体液中的溶解。目前,可以使用几种技术来解决该问题,例如,无定形固体分散体、共晶体、成盐或环糊精络合物。
环糊精是6、7或8个重复单元的环状低聚糖化合物,该重复单元具有增加的疏水腔直径。取决于重复单位的数量,它们可以分别被指定为α、β或γ。环糊精可以例如通过淀粉的酶促转化来制备。它们的外部基团是亲水的并且可以是化学改性的,而它们的内腔可以是疏水性的,这允许包含亲脂性分子,例如水溶解度低的活性药物成分。
在环糊精内腔中包含水溶解度低的活性药物成分,或形成环糊精/活性药物成分络合物,可导致水溶解度高的化合物并因此在体液中具有更高的生物利用度。
关于环糊精克服当前溶解度/生物利用度问题的优点和应用的更详细描述可以在文献的其他地方找到,即在出版物(“ciclodextrinas:comoadjuvantetecnológicoparamelhorarabiodisponiblidadedefármacos”,limaguedes等,2008年)中找到。一般而言,本综述报告了多年来使用环糊精作为药用辅料的重要性,它详细描述了环糊精的主要化学衍生物,它们的络合机制,以及它们的生物制药和毒理学意义。
用于形成环糊精/活性药物成分络合物的最常用方法的特征在于以下方法:通常将环糊精溶解在水中,然后将固体形式的活性药物成分加入到先前的溶液中。例如,通过使用搅拌和/或加热,所述活性药物成分部分地溶解在水中,并且然后与环糊精形成络合物,从而允许其他活性药物成分在溶液中溶解。为了获得粉末形式的络合物,例如,需要过滤或沉淀然后干燥的步骤。
文献中提出的方法有几个问题和缺点,即大量的用水量,由于形成的络合物数量少而产率低,并且处理时间长,需要24小时至172小时才能达到最大络合浓度,因此,超过活性药物成分在溶液中的稳定时间。据估计,由于生产量低,这些缺点使得环糊精的工业用途不那么吸引人。此外,这种方法的规模扩大通常具有挑战性和繁琐性。在美国专利6884885b2,2005中,作者声称通过增加环糊精浓度来解决第一个问题,但没有给出提供剩余问题的解决方案的其他指示。
在美国专利5646131和许多其他科学论文中,使用一种或多种亲水聚合物来增加所形成的络合物的浓度。这使得该方法更加昂贵,并且在最终粉末中存在游离聚合物,这被认为是不利的。
在专利申请us2003/0012774a1和wo2008052410a1中,使用均质化方法,并且提及通过均质化引入能量以增加辅酶q10在γ-环糊精中的络合。在两个专利申请中,辅酶q10以固态形式加入,这需要溶解和随后络合。用在均质化的能量用于将辅酶q10溶解到环糊精溶液中(即通过降低粒径的方式)。
在专利wo2015114320中,作者要求通过以下方式控制api和/或赋形剂的粒度和粒度分布的新的可扩展方法:在溶剂混合物中制备悬浮液,其中所述api和/或赋形剂部分地溶于所述溶剂的一种中;使用例如均质化方法减小所述悬浮液中存在的颗粒的尺寸;使所述悬浮液老化;并通过除去所述溶剂混合物来阻止老化。尽管老化步骤显示出是控制粒度分布的关键,但是对于形成api包合络合物,该步骤可能是不利的,因为所述络合物能够快速解离。一般来说,环糊精/api络合物的形成和解离的速率是相似的,并且它们的半衰期在溶液中只有几千秒,这意味着所述络合物不断形成和解离(limaguedes等人,2008年)。
在出版物(“具有高压均质化方法的蛇床子素羟丙基-β-环糊精包合物的物理化学表征(physicochemicalcharacterizationsofostholehydroxypropyl-β-cyclodextrininclusioncomplexeswithhigh-pressurehomogenizationmethod)”中,刘等人,2010年)中报道了使用微流化系统与活性药物成分蛇床子素(osthole)的羟丙基-β-环糊精进行络合。在这种情况下,使用高压均化器,然后进行过滤步骤和干燥步骤。这种方法比本发明效率低,因为它包括更多的工艺步骤,在高压均化器之前的混合物,在高压均化中的3个循环和在干燥之前的过滤。
在上述出版物中,活性药物成分在环糊精溶液中具有低溶解度,但由于存在环糊精,作者仍尝试在这样的条件下溶解活性药物成分。这被认为是耗时且浪费能源。也没有任何引用的出版物提供连续的过程,这是该过程经济的重要特征。
在专利us6555139b2、us2002/0086061a1、us2003/0091627a1中,作者借助于非溶剂、水与溶解的环糊精使用微流化系统来磨制活性药物成分。这些出版物的目的不是使活性药物成分络合,而是减小其粒径。在这些情况下,活性药物成分不会失去其固体形式,而在本发明中,自发的粒径减小对于活性药物成分与环糊精的络合是重要的。在这些出版物中,环糊精仅用于赋予液体某些性质而不用于络合api。
在出版物“阳离子-β-环糊精:透明质酸-金刚烷主体:客体pdna纳米粒子的微流体组装(microfluidicassemblyofcationic-β-cyclodextrin:hyaluronicacid-adamantanehost:guestpdnananoparticles)”,kulkarni等人,2013中,作者使用微流体反应器混合所有组分并使用闪光沉淀产生络合物的固体纳米颗粒。本发明认为获得络合物的溶液是有利的,因为它可以以液态形式用于可注射系统,并且任选地也可以干燥以获得适于口服递送的颗粒。
本发明的目的是提供一种环糊精和活性药物成分之间改进的络合方法,而没有已知方法的缺点,并且其可以在更短的时间内、以更高的络合物浓度、更低的溶剂量获得所需的络合物,有可能增加活性药物成分/环糊精的比例,以及能量输入以自发方式发生。所有这些因素都有助于所述方法的整体经济性。
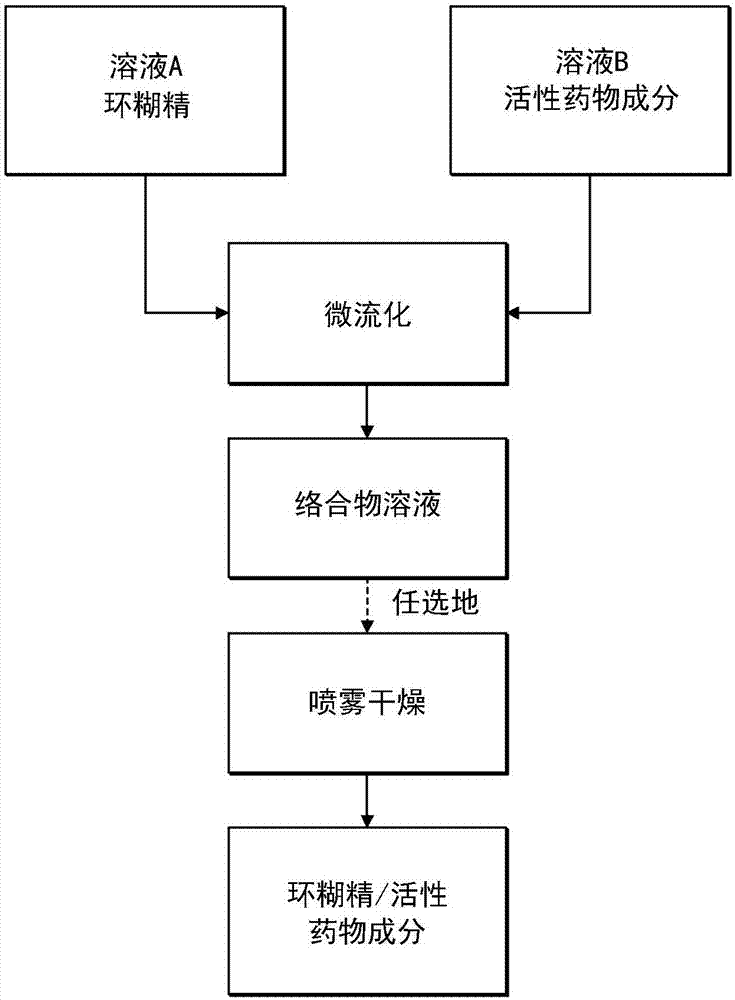
与目前现有技术不同,本发明提供了借助于微流化系统获得络合物的连续方法,该络合物由溶解在合适的溶剂中的环糊精和活性药物成分组成,该微流化系统具有高水平的混合和自发的热生成,这令人惊讶地允许在更短的时间内以高络合效率获得络合物,并且其可任选地随后喷雾干燥以获得固体材料。
技术实现要素:
本发明描述了连续或非连续方法,特别是在减少的时间内获得环糊精/活性药物成分络合物。
根据本发明,其提供制备至少一种环糊精和至少一种活性药物成分的络合物的方法,该方法包括以下步骤:
a.制备包含至少一种环糊精和至少一种溶剂的第一溶液(溶液a);
b.制备包含至少一种溶解的、部分溶解的或悬浮的api的第二溶液(溶液b);
c.通过微流化系统混合所述溶液a和溶液b,以产生至少一种所述络合物的溶液和/或悬浮液;
d.将所述溶液和/或悬浮液分离和/或任选地将其干燥;和
e.任选地收集粉末形式的络合物。
因此,本发明涉及活性药物成分(api)的增加的生物利用度,其优选通过使用微流化系统连续形成环糊精和活性药物成分的络合物来实现。或者,该方法还可以与喷雾干燥组合以分离粉末形式的络合物。更具体地,本发明涉及连续或不连续的络合方法,其中第一环糊精溶液通过将微流化与活性药物成分(api)或固体形式的活性药物成分(api)的第二溶液、部分溶液或悬浮液组合,第一溶液和第二溶液(或部分溶液、悬浮液、固体等)为可混溶或不混溶。本发明还涉及获得这种络合物的时间减少以及喷雾干燥步骤之后的络合步骤的组合,这可以使本发明成为具有高络合效率的连续过程,其中所获得的包含络合物的粉末具有高载药量。因此,本发明在减少处理时间方面具有益处。
该方法可以是分批法或连续法。
溶液a优选包含至少一种环糊精,该至少一种环糊精包括任何取代基和任何腔体尺寸,和/或聚合物药物赋形剂。所述环糊精可以是,例如,α-环糊精、β-环糊精、γ-环糊精、磺丁基醚-β-环糊精、羟丙基-β-环糊精、甲基-β-环糊精和/或麦芽糖基-β-环糊精中的一种或多种。特别优选的环糊精是磺丁基醚-β-环糊精。
溶液a优选地包括以下溶剂中的至少一种:水、乙醇、甲醇、异丙醇、二氯甲烷丙酮、甲基乙基酮、四氢呋喃、二甲基亚砜、二甲基甲醛或二甲基乙酰胺。
溶液a中环糊精的浓度范围优选地为从1%到50%(w/w)。
如果需要,聚合物药物赋形剂可以存在于溶液a中,例如浓度为从1%到20%(w/w)。
溶液a的ph的范围通常为从1到14,优选的ph为从6至8。
在本发明的方法中,溶液a优选如下制备:使用带有搅拌的夹套反应器,向该反应器中加入至少一种溶剂并且将至少一种环糊精加入同一反应器和/或将至少一种聚合物药物赋形剂加入同一反应器中,通常随后进行ph调节。添加上述组分的顺序不受限制,并且可以是任何方式。
溶液b优选地包括至少一种溶解在一种或多种溶剂中、部分地溶解在一种或多种溶剂中、或悬浮在一种或多种溶剂中的api。优选地,api或其药学上可接受的衍生物具有至少一个选自以下的官能团:硫醚、醇、硫醇、醛、酮、硫酮、硝酸酯、磷酸酯、硫代磷酸酯、酯、硫酯、硫酸酯、羧酸、膦酸、磺酸、酰胺、伯胺、仲胺、氨、叔胺、亚胺、硫氰酸酯、氰胺、肟、腈、重氮、有机卤化物、硝基、s-杂环、噻吩、n-杂环、吡咯、o-杂环、呋喃、环氧化物、过氧化物、异羟肟酸、咪唑和吡啶。
优选的api的实例包括但不限于难溶的活性化合物。难溶性化合物的实例包括但不限于:抗真菌剂如伊曲康唑或相关药物,例如氟康唑、特康唑、酮康唑和沙康唑;抗感染药物,例如灰黄霉素和相关化合物(例如灰绿霉素);抗疟疾药物(例如阿托伐醌);蛋白激酶抑制剂,例如阿法替尼、阿西替尼、博舒替尼、西妥昔单抗、克唑替尼、达沙替尼、厄洛替尼、fostamatinib、吉非替尼、依鲁替尼、伊马替尼、zemurasenib、拉帕提尼、乐伐替尼、木利替尼或尼罗替尼;免疫系统调节剂(例如环胞菌素);心血管药物(例如地高辛和螺内酯);布洛芬;甾醇或类固醇;来自包括以下药品的药物:达那唑、阿昔洛韦、氨苯砜、茚地那韦、硝苯地平、硝基呋喃、苯丙噻嗪、利托那韦、沙奎那韦、磺胺甲恶唑、丙戊酸、甲氧苄啶、乙酰唑胺、硫唑嘌呤、异丙酸、萘啶酸、奈韦拉平、吡喹酮、利福平、阿苯达唑、阿米替林、蒿甲醚、苯芴醇、氯丙嗪、环丙沙星、氯法齐明、依法韦仑、洛匹那韦、叶酸、格列本脲、氟哌啶醇、伊维菌素、甲苯咪唑、氯硝柳胺、噻嘧啶、乙胺嘧啶、视黄醇维生素、磺胺嘧啶、柳氮磺胺吡啶、三氯苯达唑和桂利嗪。
溶液b优选地包含以下溶剂中的至少一种:水、乙醇、甲醇、异丙醇、二氯甲烷、丙酮、甲基乙基酮、四氢呋喃、二甲基亚砜、二甲基甲醛或二甲基乙酰胺。
在本发明中,溶液b中的一种或多种溶剂可与溶液a中的那些溶剂相同或不同,且如果不同,则所述溶剂可互相溶混或互不溶混。
溶液b中的api的浓度优选地在0.01%到100%(w/w)范围内。
溶液b通常是使用带搅拌的夹套反应器通过将至少一种溶剂添加到所述反应器和至少一种api来制备。添加组分的顺序不受限制且可以是任何方式。
在一个方面,如果需要,例如可由正位移设备(例如,基于螺杆的进给系统或气动系统)连续地进给固体api。
在一个优选方面,优选地使用料斗进给所述固体api。
微流化优选地发生在包含至少一个增压泵的系统中,所述系统可将与另一种混溶或不混溶的液体混合的液体,或可替代地与固态化合物混合的液体混合并排出到至少一个微通道或微反应器中。如果需要,可使用两个或多个微通道或微反应器。
优选地,微通道或微反应器是适当地具有等于或小于1000微米的横向尺寸的连续流反应器。该横向尺寸可等于或小于200微米。
所述方法的动水压力优选地在1巴到1500巴的范围内,且可适当地在250巴到1000巴的范围内。
在本发明的一个优选方面,溶液a与溶液b或固体api的进料流量比在约0.1到约10kg/kg的范围内。溶液a与api的添加量的进料流量比可在0.01到10kg/kg的范围内。
考虑到所涉及的组分,该方法的温度可以是任何合适的温度,且适当地在0℃到90℃的范围内。
将络合物从络合物溶液和/或悬浮液分离可通过任何方式来完成,但优选地包括一种或多钟干燥技术。一种特别适合的干燥技术包括喷雾干燥。
在本发明方法的方法中,将络合物从络合物溶液和/或悬浮液分离可为分批法或连续法。
在一优选方面,所述络合物是以粉末形式分离出的。
应理解,通过本发明的方法产生的络合物可用于医药中,例如作为药物组合物的一部分。本发明因此提供此用途,或替代地一种方法,其特征在于所获得的溶液和/或悬浮液或粉末形式的络合物用于药物目的。
在一优选方面,本发明提供一种用于制备至少一种环糊精和至少一种活性药物成分的络合物的方法,所述方法包含以下步骤:
a)利用至少一种环糊精在一种或多种溶剂中制备溶液a;
b)利用至少一种溶解或悬浮的api制备溶液b。所述溶剂可与溶液a相同或不同,且如果不同,则可以是可混溶或不混溶的。替代地,所述一种或多种活性药物成分通过正位移设备,优选地料斗和螺旋进料器,以固态添加;
c)使用包含增压泵的微流化系统连续混合所述溶液a与溶液b或固态的api,所述增压泵将所有组分抽出并混合到产生气穴的高压室且随后将混合物排出到至少一个微通道中,从而形成络合物溶液;
d)任选地通过例如结晶、过滤或干燥(优选喷雾干燥)的分离方法将所述络合物与溶液连续或不连续地分离。
本发明提出减少形成环糊精与至少一种活性药物成分之间的络合物的络合时间,由于高水平的混合与自发热产生,使得使用本发明连续制造环糊精络合物,出乎意料地产生高浓度的所需络合物。当例如与喷雾干燥组合使用时,可能获得包含相同络合物的粉末。
本发明还提出一种以少于已知工艺的时间获得具有较高浓度的与环糊精络合的活性药物成分的粉末的方法。
优选地,本发明能够连续生产环糊精/活性药物成分络合物且通过使用喷雾干燥进行连续分离。
本发明包括一种微流化系统。该微流化系统通常由增压泵组成,所述增压泵可将与其它混溶或不混溶的液体混合的液体或替代地与固体化合物混合的液体混合并排出到微通道的组合中。所述微通道通常是具有1000μm的横向尺寸的连续流反应器。
优先地,本发明使用两种不同装置:
装置a包含:
a)用来制备溶液a的反应器;
b)用来将固体api排放到反应器a)的例如料斗和螺旋进料器的正位移设备;
c)增压泵;
d)至少一个微通道的组合。
任选地,用来收集粉末状络合物的喷雾干燥器和旋风器;
装置b
a)用来制备溶液a的反应器;
b)用来制备溶液b的反应器;
c)增压泵;
d)至少一个微通道的组合。
任选地,用来收集粉末状络合物的喷雾干燥器和旋风器。
附图说明
图1示出本发明方法的示意性流程图。
图2示出装置a的示意图,其中a)是混合反应器,b)是正位移设备,c)是增压泵,d)是至少一个微通道的组合,e)是干燥室,以及f)是旋风器。
图3示出装置b的示意图,其中a)是混合反应器,b)是第二混合反应器,c)是增压泵,d)是至少一个微通道的组合,e)是干燥室,以及f)是旋风器。
图4示出所形成的络合物的浓度与时间的表示。虚线表示实施例1方法。断续线表示实施例2方法。实线表示实施例3方法。
具体实施方式
本发明提出一种用于环糊精与至少一种活性药物成分络合后有干燥步骤的连续或不连续方法。干燥步骤可例如是喷雾干燥方法,但可使用其它合适的干燥方法。
更具体地,本发明提出一种用于通过将包含至少一种溶解环糊精的溶液a与包含至少一种溶解或者悬浮api的溶液b或替代的由正位移设备(优选地配备有螺旋进料器的料斗)进给的固体api混合而形成流b来使环糊精与至少一种活性药物成分络合的连续或不连续(间断)方法。然后使用增压泵将流a和流b抽出并将其混合到产生气穴的泵高压室且随后将所述混合流排放到产生高剪切混合的至少一个微通道中,通过气穴研磨api,由于摩擦自然地产生热,且出人意料地产生包含高浓度络合物的络合物溶液。如果与喷雾干燥步骤合并,则连续或不连续地干燥所获得的络合物溶液,从而产生粉末形式的环糊精与活性药物成分的络合物。
溶液a包含具有优选地选自以下的一种或多种溶剂的溶液:水、乙醇、甲醇、异丙醇、二氯甲烷、丙酮、甲基乙基酮、四氢呋喃、二甲基亚砜、二甲基甲醛或二甲基乙酰胺。溶液a还包含一种或多种溶解环糊精或经取代环糊精。可使用任何环糊精或经取代环糊精,且优选化合物包括例如α-环糊精、β-环糊精、γ-环糊精、磺丁基醚-β-环糊精、羟丙基-β-环糊精、甲基-β-环糊精和/或麦芽糖基-β-环糊精。任何一种或多种合适的药物赋形剂也可以例如1%(w/w)到20%(w/w)的浓度添加到溶液a。所述一种或多种环糊精的浓度优选地是1%(w/w)到50%(w/w)。溶液a具有1到14范围内的ph值,优选地6到8。溶液a可例如如下来制备:使用带搅拌的夹套反应器,将溶剂添加到该反应器中且将一种或多种环糊精添加到同一反应器,之后进行ph调整。添加顺序不受限制,且以任何方式完成。
溶液b包含使用优选地选自以下的一种或多种溶剂的溶液或悬浮液:水、乙醇、甲醇、异丙醇、二氯甲烷、丙酮、甲基乙基酮、四氢呋喃、二甲基亚砜、二甲基甲醛或二甲基乙酰胺。溶液b还包含至少一种可溶解、部分溶解或悬浮在溶剂中的活性药物成分(api)。所述api优选地以0.01%(w/w)到100%(w/w)范围内的浓度存在。替代地,可使用固体形式的活性药物成分。可例如使用带搅拌的夹套反应器制备溶液b,其中所述一种或多种溶剂和至少一种活性药物成分添加到反应器。添加顺序不受限制,且以任何方式完成。
替代地,所述至少一种固体形式的api可直接添加到混合物中。优选地,该固体形式的api由优选地包含料斗或螺旋进料器的正位移设备,或由气动装置添加。
根据本发明,溶液a与溶液b之间存在最佳进料流量比。因此,例如,到微流化系统的溶液a的进料流可以是到微流化系统的溶液b的进料流的0.1到10倍范围内。也可使用0.5到5,或0.5到3,或1到3的比率(按照以上定义)。
替代地,在本发明中,溶液a与固体api的添加量的进料流量比之间存在最佳比率。因此,例如,到微流体设备的溶液a的进料流可以是固体活性药物成分的添加量的0.01到10倍范围内。也可使用0.1到10,或0.1到5,或0.5到3,或1到3的比率(按照以上定义)。
根据本发明,所有化合物的混合和络合在微流化系统中进行。“微流化”是本领域技术人员能理解的术语。术语“微反应”是指一种涉及到微反应器、微混合器、微通道或任何其他微流体领域内包括的部件内的物理和/或化学反应的技术。术语“微流化”涵盖了通过这些微通道的连续流体处理,其中涉及到在介观和微观混合范围内的高剪切、气蚀和均匀混合。例如,微流化系统包括增压泵,该增压泵将流a和溶液b或至少一种固体形式的api连续地或非连续地(即不连续地)吸入泵室中,然后推动流的混合物通过至少一个微通道,由此产生高压、自发生热、摩擦、气蚀、高雷诺数下的高剪切混合以及研磨(如果api是固体形式的话)。我们惊讶地发现,在加压通路的末端获得了高浓度的络合物。络合物的形成包括至少一种api的溶解,并因此包含在至少一种环糊精的空腔中。微通道的数量不受限制并且可以是至少一个,但优选的范围为1至10个。微通道的横向尺寸小于约1000μm,并且优选地等于或小于约200μm。泵形成1巴至1500巴的压力(我们能给出一些优选的子范围吗?)并且混合液体的温度自发地升高,这表明效率因为干燥混合液体所消耗的能量更少而得到了提高。在该方法结束时,获得了具有高浓度的环糊精/活性药物成分络合物的溶液,所花费的时间比之前报道的要短得多。
如果需要,可以将所得溶液连续地或非连续地进料到喷雾干燥器中。例如,喷雾干燥器可以配有将络合物溶液雾化成液滴的喷嘴以及流速为1至2000kg/h的流过干燥气体,并且借助于0℃至200℃的温度,对络合物溶液液滴进行干燥,所形成的固体颗粒在旋风分离器中进行收集。在喷雾干燥操作结束时,获得环糊精/活性药物成分络合物的含量较高的粉末。
本发明可以是连续法或非连续法。例如,在非连续实施方案中(装置a),溶液a和溶液b或固体api可以在反应器中预混合,微流化,然后再循环回到产生络合物溶液的相同反应器中。可选地,对来自本实施方案的所述络合物溶液进行干燥,例如采用喷雾干燥或冷冻干燥的方式来进行干燥。
在连续实施方案中(例如装置b),本发明公开了一种其中对溶液a和溶液b或固体api实施微流化以产生络合物溶液的方法。可选地,将所述络合物溶液连续进料到连续干燥器中,例如喷雾干燥器。
由于每次处理时间所获得的固体络合物的量有所增加,因此,本发明具有高产量。之所以能实现此目的是因为以下三种主要效果的结合:各组分在溶液a和溶液b中的溶解时间缩短;混合、研磨和自发温度(提高了络合物溶液中的络合物浓度)的水平上升使得络合时间缩短;最后,可选地使所述络合物溶液连续地进入喷雾干燥,得到固体络合物。
实施例
实施例1:
将60克水加入带有搅拌的反应器中。向此反应器中加入40克的磺丁基醚-β-环糊精。连续地搅拌悬浮液,直至形成澄清溶液。然后,将1克活性药物成分(在这种情况下使用伊曲康唑作为模型药物)加入到所述溶液中,并立即设定好计时器。所形成的悬浮液在25℃下连续搅拌。每天过滤2克所述悬浮液并通过hplc对其进行分析,以确定出络合物浓度。图4中示出了结果(虚线)。
实施例2:
将60克水加入带有搅拌的反应器中。向此反应器中加入40克的磺丁基醚-β-环糊精。连续地搅拌悬浮液,直至形成澄清溶液。在使用装置a的情况下,将10克活性药物成分(伊曲康唑)加入到所述溶液中,并立即设定好计时器。所形成的悬浮液在550巴的压力下在室温下进料到增压泵,持续1小时。每隔10分钟过滤来自悬浮液的2克样品并通过hplc对其进行分析,以确定出络合物浓度。1小时后,对溶液实施喷雾干燥,得到粉末状物质。图4中示出了结果(虚线)。
实施例3:
将60克水加入带有搅拌的反应器中。向此反应器中加入400克的磺丁基醚-β-环糊精。连续地搅拌悬浮液,直至形成澄清溶液。在使用装置a的情况下,将10克活性药物成分(伊曲康唑)加入到所述溶液中,并立即设定好计时器。所形成的悬浮液在550巴的压力下在室温下进料到增压泵,持续3小时。每隔10分钟过滤来自悬浮液的2克样品并通过hplc对其进行分析,以确定出络合物浓度。3小时后,对溶液实施喷雾干燥,得到粉末状物质。图4中示出了结果(实线)。
实施例4:
将60克水加入带有搅拌的反应器中。向此反应器中加入40克的磺丁基醚-β-环糊精。向另一个反应器中加入90克的二氯甲烷。向此反应器中加入10克的活性药物成分(伊曲康唑)。连续地搅拌这两种溶液,直至形成澄清溶液。在使用装置b的情况下,将两种溶液进料到增压泵,其中溶液a相对于溶液b的进料流量比为10千克/千克,并且在50℃的温度下产生1000巴的压力。对所得溶液取样以确定出络合物浓度。之后对溶液实施喷雾干燥,形成粉末状物质。
- 还没有人留言评论。精彩留言会获得点赞!