治疗物质滥用障碍用组合物和方法与流程
本发明是在国家药物滥用研究所(nationalinstituteondrugabuse)授予的美国公共卫生基金1r01da030932-01的政府支持下完成的。
本发明涉及用于治疗如下各种病状和病症的方法,包括神经精神病症,如成瘾、焦虑、抑郁、精神分裂症和相关病状(例如失眠),并且更一般地涉及制备和使用靶向神经和内分泌系统内不同组织的药物制剂的方法。
背景技术
虽然几十年来科学家一直在研究精神运动刺激剂奖励的神经生物学,但仍然没有fda批准的用于可卡因或甲基安非他命(methamphetamine)滥用的治疗。以前的实验室研究关注应激、随后的下丘脑-垂体-肾上腺(hpa)轴活化和精神运动刺激剂强化之间的关系达近30年。这种研究使得已经开发出低剂量皮质醇合成抑制剂甲吡酮与苯并二氮杂卓奥沙西泮(oxazepam)的组合作为可卡因和其它物质使用障碍的潜在药理学治疗。事实上,已经进行了试验性临床试验,展示这种组合可以减少可卡因渴求和可卡因使用。这种作用的潜在假设是,甲吡酮与奥沙西泮的组合通过降低hpa轴内的活动减少了可卡因寻求和摄取。即便如此,持续减少可卡因摄取和寻求的甲吡酮与奥沙西泮组合的剂量并不能可靠地改变血浆皮质酮(或试验性临床试验中的皮质醇)。此外,随后的研究展示,这种药物组合在肾上腺切除的大鼠中是有效的,从而表明这些作用必须在肾上腺水平的上方介导。正在形成的假设是,甲吡酮与奥沙西泮的组合通过增加内侧前额叶皮质和杏仁核中的神经活性类固醇、最显著的是四氢脱氧皮质酮的水平而产生其作用。
在过去几年中,对精神运动刺激剂强化中应激与随后的下丘脑-垂体-肾上腺(hpa)轴的活化之间的复杂关系进行了大量研究(goeders,2002,2007;majewska,2002;winhusen和somoza,2001;sarnyai等人,2001)。在这种情形下,已经研究了减少hpa轴活动的药物对可卡因自身给药以及药物和线索诱导的已消退可卡因寻求的恢复的作用(goeders,2004,2007)。
这个领域的早期工作研究了苯并二氮杂卓受体激动剂对大鼠静脉内可卡因自身给药的作用。鉴定这类药物不仅是因为它们属于用于治疗焦虑的最广泛处方药物(uhlenhuth等人,1995;baldessarini,1996),还因为这些药物可以降低血浆皮质酮(keim和sigg,1977)、皮质醇和acth(meador-woodruff和greden,1988;torpy等人,1993)并且减少可卡因诱导的血浆皮质酮增加(yang等人,1992)。最初报道用利眠宁(chlordiazepoxide)预处理显著降低了静脉内可卡因自身给药(goeders等人,1989)。当单位剂量的可卡因增加时,这种作用减弱,表明利眠宁降低可卡因作为强化剂的功效。然而,由于这些药物摄入的减少可能是由于大鼠反应能力的非特异性破坏,因此进行了另一项研究,其中在大鼠中测试了另一种苯并二氮杂卓受体激动剂阿普唑仑(alprazolam),所述大鼠在静脉内可卡因提供和食物强化的多重方案下反应(goeders等人,1993)。最初,阿普唑仑减少了通过食物和可卡因两者维持的反应。然而,尽管在后来的测试期间对于阿普唑仑对食物维持反应的镇静作用迅速产生耐药性,但没有观察到对于阿普唑仑减少可卡因自身给药的能力的耐药性,从而表明苯并二氮杂卓的作用可能来自对可卡因强化的特异性作用而不是对反应的非特异性作用。
还有一些研究关注皮质酮合成抑制剂对可卡因自身给药的作用。甲吡酮阻断皮质酮产生中的11β-羟基化反应而降低该激素的血浆浓度(haleem等人,1988;haynes,1990)。用甲吡酮预处理使大鼠的可卡因自身给药和血浆皮质酮产生显著剂量相关的下降(goeders等人,1996)。然而,由于再次不明确这些作用是特异于可卡因强化还是对大鼠反应能力的非特异性作用的结果,因此通过使用食物提供和可卡因自身给药的多重交替方案进行了另一个实验来解决这个问题,这次在用酮康唑预处理后进行。酮康唑是一种具有广谱活性和低毒性的口服抗霉菌剂(sonino,1987;thienpont等人,1979),其还抑制肾上腺皮质类固醇合成中的11β-羟基化和18-羟基化步骤(engelhardt等人,1985)。在这些实验中,允许大鼠在每天2小时训练期期间经受交替的15min周期的食物强化和可卡因自身给药。用酮康唑预处理减少了可卡因自身给药而不影响食物强化的反应,从而表明皮质酮合成抑制剂在不产生非特异性运动作用的剂量下降低可卡因强化。
因此,已经展示苯并二氮杂卓受体激动剂和皮质酮合成抑制剂减少了可卡因自身给药。然而,这两类药物都有潜在的副作用,所述副作用可以限制所述药物在可卡因成瘾治疗中的适用性。例如,通常不建议苯并二氮杂卓作为可卡因依赖的治疗选择,因为这些药物具有滥用的可能性(chouinard,2004;lilja等人,2001;o'brien,2005),引起了对使用这些药物可能造成二次依赖的担忧(wesson和smith1985)。皮质酮合成抑制剂具有产生尤其肾上腺功能不全的可能性,这也尤其可以限制这类药物的效用。然而,可以通过减少剂量来减轻由这两类药物产生的副作用的发生。具体地,通过将经由不同机制影响hpa轴活动的药物组合并以单独施用时无作用的浓度递送这些药物,可以使其潜在的毒性和不想要的副作用达最小,同时仍然减少可卡因的摄入。如以下实施例中所述,通过使用甲吡酮与奥沙西泮的组合证实了这个理论。
技术实现要素:
本发明部分基于以下发现:某些类型的治疗剂可以组合使用以治疗各种神经精神病症和相关病症,包括成瘾(例如,对物质如药物或对活动如赌博成瘾)。更具体地,根据本发明,甲吡酮与奥沙西泮的组合已经显示在人类中是安全且良好耐受的,因此具有安全且有效治疗人类的物质滥用障碍和成瘾的强大潜力。此外,这些药剂有潜力被安全地用于治疗人类的进食障碍;抑郁;破坏性行为障碍(例如,注意力不足障碍,如注意力不足和多动障碍(adhd));精神分裂症;焦虑(例如,在创伤后应激障碍的情形下经历的焦虑);睡眠障碍;和/或相关的或所引起的病状。本发明还包括可用于治疗或预防肥胖或各种进食障碍的组合物和方法。所述组合物还可以用于治疗或预防失眠,所述失眠可以独立地发生或与应激相关病状或应激相关障碍(例如焦虑)有关。更一般地,这些病状可以被描述为与以下相关的病状:皮质醇增多、下丘脑-垂体-肾上腺(hpa)轴(例如促肾上腺皮质激素(acth)的调节改变)或前额叶皮质内的其它活动和/或交感神经系统中的过度活动。
因此,本发明的特征在于药物组合物和能够制备以及向患者施用(例如开药和自身给药)所述药物组合物的方法。本文所述的治疗剂可以配制成单一制剂(例如,单一片剂、胶囊等,其可以被设计来产生持续和控制释放)并口服施用。然而,本发明不限于此,并且下文进一步描述了用于组合和施用所述治疗剂的示例性替选方案(例如,可以静脉内施用溶液)。
无论精确的配制物或配置如何,组合物都可以包括至少一种靶向下丘脑-垂体-肾上腺(hpa)轴的活性成分和至少一种靶向前额叶皮质(例如,通过靶向前额叶皮质中的gabaa受体)的活性成分。更具体地,所述组合物可以包括至少一种第一活性剂,其抑制促皮质素释放激素(crh);抑制促肾上腺皮质激素(acth);和/或抑制皮质醇。例如,所述药剂可以降低crh刺激从垂体腺释放acth的能力;降低acth刺激从肾上腺释放皮质醇的能力,或抑制皮质醇合成、分泌或活性。例如,虽然本发明的组合物不限于通过任何特定机制发挥其作用的组合物,但是抑制皮质醇活性的药剂可以通过与皮质醇竞争糖皮质激素受体结合和/或阻断经由糖皮质激素反应元件的下游事件如受体活化、二聚或转录信号传导来发挥作用。这些药剂也可以是与另一类肾上腺皮质类固醇受体如盐皮质激素受体结合和/或抑制盐皮质激素受体结合后的下游事件的药剂。
所述组合物还可以包括至少一种第二活性剂,所述第二活性剂通过以下方式靶向前额叶皮质,例如:增加γ-氨基丁酸(gaba)的表达或活性;模拟gaba;抑制前额叶皮质中的gaba代谢;和/或以其它方式刺激前额叶皮质中的gaba信号传导。如上所述,组合物可以通过将这些第一和第二药剂本身在例如延长释放制剂中物理组合(例如以混合物或悬浮液形式)来含有所述药剂。在其它实施方式中,组合物可以通过共享包装而组合(例如,含有第一活性剂的片剂和含有第二活性剂的片剂可以组合在单个泡罩包装中,对所述泡罩包装任选地标记以指示一周的天数和/或一天的时刻)。用于静脉内施用的溶液可以类似地包装,其中一种溶液含有第一药剂,并且一种溶液含有第二药剂,并提供同时或依序施用的说明。所述组合物还可以包括至少一种第二活性剂,所述第二活性剂通过以下方式靶向前额叶皮质,例如:增加γ-氨基丁酸(gaba)的表达或活性;模拟gaba;抑制前额叶皮质中的gaba代谢;和/或以其它方式刺激前额叶皮质中的gaba信号传导。如上所述,组合物可以通过将这些第一和第二药剂本身在例如延长释放制剂中物理组合(例如混合物或悬浮液形式)来含有所述药剂。在其它实施方式中,组合物可以通过共享包装而组合(例如,含有第一活性剂的片剂和含有第二活性剂的片剂可以组合在单个泡罩包装中,对所述泡罩包装任选地标记以指示一周的天数和/或一天的时刻)。用于静脉内施用的溶液可以类似地包装,其中一种溶液含有第一药剂,并且一种溶液含有第二药剂,并提供同时或依序施用的说明。
在特定实施方式中,所述组合物可以包括下表第一栏中列出的一种或多种类型的药剂和第二栏中列出的一种或多种类型的药剂。
表-us-00001
这些类型的药剂中的任一种或两种可以与抑制交感神经系统中的活动的药剂(例如β-阻断剂,如普萘洛尔(propranolol)
在特定实施方式中,抑制crh的药剂可以与刺激前额叶皮质中的gaba的药剂以及抑制交感神经系统中的活动的药剂组合;抑制acth的药剂可以与刺激前额叶皮质中的gaba的药剂以及抑制交感神经系统中的活动的药剂组合;抑制皮质醇的药剂可以与刺激前额叶皮质中的gaba的药剂以及抑制交感神经系统中的活动的药剂组合;并且一种或多种与肾上腺皮质类固醇受体结合的药剂可以与抑制交感神经系统中的活动的药剂组合。在这些示例性实施方式中的任一个中,所提及药剂可以是本文所述的药剂(例如刺激gaba的药剂可以是直接或间接刺激前额叶皮质中的gaba的药剂;模拟前额叶皮质中的gaba的药剂(例如gaba受体(例如gabaa)激动剂);或抑制gaba代谢的药剂)。
gaba是一种抑制性神经递质,其在受体结合后使受抑制的神经元超极化。这种结合直接或间接地打开了氯通道和钾通道。活化的离子型受体本身是离子通道,而代谢型受体是通过中间g蛋白活化离子通道的g蛋白偶联受体。任一类型的受体都可以被用于模拟gaba的药剂活化,从而靶向前额叶皮质。其它药剂可以通过增加gaba合成起作用。例如,可以将编码合成酶l-谷氨酸脱羧酶的核酸或其生物学活性片段或其它突变体施用于可能受益于本文所述方法的患者(例如已经证实或已经诊断为患有成瘾的患者(适合接收治疗的其它患者在本文别处描述))。
在其它实施方式中,治疗性组合物可以是以下药剂中的至少两种或三种(例如两种、三种或四种)的组合:抑制crh的药剂、抑制acth的药剂、抑制皮质醇(或结合肾上腺皮质激素受体)的药剂、直接或间接刺激前额叶皮质中的gaba的药剂、模拟前额叶皮质中的gaba的药剂或抑制gaba代谢的药剂以及抑制交感神经系统中的活动的药剂。
除非上下文另外指示,否则术语“药剂”广泛地用于指以临床上有益的方式影响靶分子(例如与其结合的配体或受体)或者脑或内分泌系统的靶区域(例如在患者暴露于一种或多种条件环境线索后抑制hpa轴活化)的任何物质。例如,化学化合物如甲吡酮
尽管下文进一步描述了适用于本发明组合物的药剂,但在此应注意,可抑制hpa中的crh的药剂包括抑制crh表达的药剂(例如核酸);通过参与负反馈循环抑制crh产生或分泌的药剂;特异性结合并抑制crh的抗体;crh受体拮抗剂(例如蛋白质,包括抗体,其结合crh受体并抑制信号转导或在细胞内起作用以抑制通常响应于crh受体结合而产生的第二信使);抑制crh的表达、分泌或活性或crh受体的化学化合物(例如小分子)(例如,抑制crh结合垂体中同源受体的能力的化合物);以及促进crh代谢的药剂。如所述,其它药剂可以抑制acth。例如,本发明的组合物可以包括抑制acth表达的药剂(例如核酸);通过参与负反馈循环抑制acth产生或分泌的药剂;特异性结合并抑制acth的抗体;acth受体拮抗剂(例如结合acth受体并抑制信号转导或在细胞内起作用以抑制通常响应于acth受体结合而产生的第二信使的蛋白质);抑制acth的表达、分泌或活性或acth受体的化学化合物(例如,抑制acth结合肾上腺中同源受体的能力的化合物);以及促进acth代谢的药剂。
抑制crh的药剂包括[met18,lys23,glu27,29,40,ala32,41,leu33,36,38]crf9-41,其缩写为α-螺旋crf(9-41)并具有序列asp-leu-thr-phe-his-leu-leu-arg-glu-met-leu-glu-met-ala-lys-ala-glu-gln-g-lu-ala-glu-gln-ala-ala-leu-asn-arg-leu-leu-leu-glu-glu-ala(seqidno:1),以及其生物学活性片段或变体(rivier等人,science224:889,1984)。另一种抑制crh的药剂是[d-phe12,nle21,38,(αmeleu37)]crf(12-41),其缩写为d-phecrf12-41,以及其生物学活性片段和变体。抑制crh的其它药剂包括
为了抑制acth,可以施用足量acth以通过反馈抑制来抑制acth,或下调acth受体。
抑制皮质醇的化学化合物包括甲吡酮、酮康唑和氨鲁米特。有用的化合物和其它药剂,包括本文中具体描述和/或本领域中以其它方式已知的那些,可以沿着hpa轴的任何点起作用来下调皮质醇的作用(即它们可以直接(例如通过结合和抑制靶标)或间接(例如通过抑制hpa轴中靶标上游的活性分子)作用于靶标(例如皮质醇))。
物质p拮抗剂和血管加压素抑制剂也可以用于本发明的组合物和方法中来抑制hpa轴内的活动。物质p是一种结合神经激肽1受体的11个氨基酸的神经肽。拮抗剂包括目前可用于化疗诱发的恶心的
增强内源性大麻素信号传导的药剂也可以用于抑制hpa轴中的活动,并且适用于本发明的组合物和方法中。这些药剂可以刺激内源性大麻素的表达或活性,或可以例如作为或模拟内源性大麻素(参见patel等人,endocrinol.145:5431-5438,2004)。虽然本发明不限于通过任何特定机制对本文所述的病症和其它病状发挥其积极作用的药剂,但应注意内源性大麻素可以抑制从垂体后叶释放血管加压素(tasker,endocrinol.145:5429-5430,2004).29-5430。外源性大麻素已显示能刺激hpa,但至少一种这种化合物cp55940反而能够减少应激诱导的hpa激素分泌(thomas等人,j.pharmacol.exp.ther.285:285-292,1998)。
直接或间接刺激前额叶皮质中的gaba的药剂可以通过直接或间接增加gaba的合成、释放或活性而发挥该作用。活性可以例如通过提高gaba与同源受体之间的相互作用来提高。有各种方法提高这种相互作用,包括增加gaba的浓度、提供受体激动剂或改变受体结合和信号转导的动力学。gaba浓度又可以通过增加gaba合成或抑制gaba代谢来增加。实际上,施用模拟gaba的药剂也增加gaba浓度。关于间接刺激,任何优选增加前额叶皮质中的多巴胺能或去甲肾上腺素能活性的药剂(例如抗抑郁药)可以间接影响(即刺激)前额叶皮质中的gaba。米氮平(mirtazapine)是抗抑郁药的一个实例,其可以用于间接刺激gaba;托莫西汀(atomoxetine)是可以类似使用的另一类药剂的一个实例。加巴喷丁(gabapentin)(neurontintm)是模拟gaba作用的药剂的实例,并且直接刺激剂包括任何苯并二氮杂卓(例如奥沙西泮
虽然下面将进一步描述剂量,但是当在本发明的组合物中使用的药剂是目前已知的并且用于治疗患者的药剂时,在组合疗法的情形下所需的至少一种药剂的剂量可以小于所述药剂目前和通常被开出的剂量。例如,当本发明的组合物包括目前用于治疗焦虑的苯并二氮杂卓时,施用于患者来治疗成瘾的化合物的量可以少于医生通常为治疗焦虑而开出的剂量。在某些情况下,本发明组合物中两种药剂的剂量将少于这些药剂的传统剂量。虽然本发明的组合物不限于具有特定优点的组合物,但使用低剂量制剂的能力可以减少副作用的发生以及与一些药剂相关的滥用可能性。在本领域中应理解,一些患者可能或多或少对特定剂量的给定药物敏感。在本发明的情况下,通常地,患者和其健康护理者可以就期望效果监测治疗,并且可以各种各样地调节剂量(例如,随着时间的推移)。
本发明组合物内化学化合物的量可以变化。例如,患者可以以限定的间隔接受约1-1000mg的给定第一药剂和1-1000mg的给定第二药剂。如果包括第三药剂,则制剂可以包括并且患者可以接受1-1000mg的第三药剂。例如,患者可以每数小时(例如约每2、4、6、8、12或24小时)、每数天(例如一天一次、每隔一天一次、每三天一次)或每数周(例如,一周一次)地进行治疗。例如,患者可以每天1-4次接受至少或约5-1500mg(例如至少或约5、10、25、50、100、200、250、300、400、450、500、550、600、650、700、750、800、850、900、1000、1250或1500mg)的第一药剂和至少或约5-500mg(例如至少或约1、5、10、20、25、30、35、40、45、50、100、200、250、300、400、450、500)的第二药剂。在这种方案下,患者可以接受至少或约10-6000mg的第一药剂(例如至少或约25-1500mg;50-1250mg;100-1250mg;100-1000mg;250-1000mg;500-1000mg;750-1000mg(例如约750mg或约1000mg)),如甲吡酮或酮康唑。在相同或不同的方案下,患者可以接受约5-100mg的第二药剂(例如约5-50mg;约5-40mg;约5-30mg;约5-20mg;约5-10mg;约10-50mg;约10-40mg;约10-30mg;约20-50mg;约20-40mg;约20-30mg;约30-50mg;或约30-40mg的第一药剂)。如所述,第二药剂可以是苯并二氮杂卓,如奥沙西泮。如所述,适当的剂量能够从可以每天或每周间隔施用的延长释放制剂随时间递送。如果使用特定制剂或装置(例如输注泵),则可以在不需要较长时间的患者干预的情况下进行施用。
药物制备物内药剂的量可以相同或不同(例如,第一药剂与第二药剂的比率可以是至少或约100:1;90:1;80:1;75:1;70:1;65:1;60:1;55:1;50:1;45:1;40:1;35:1;30:1;25:1;20:1;15:1;10:1;9:1;8:1;7:1;6:1;5:1;4:1;3:1;2:1;或约1:1)。例如,组合物可以含有约1当量奥沙西泮比上约25-50当量甲吡酮;约25-50当量酮康唑比上约1当量阿普唑仑;约25-50当量酮康唑比上约1当量奥沙西泮;约25-50当量甲吡酮比上约1当量阿普唑仑;约1当量蝇蕈醇比上约25-50当量cp-154,526;或约1当量蝇蕈醇比上约25-50当量甲吡酮。当量可以是重量单位(例如毫克)。然而,比率可以不同,其中第二药剂的量超过第一药剂的量(例如,按照本文描述的不同程度)。活性成分的相对量也可以用百分比表示。例如,相对于彼此,第二药剂的量可以是第二药剂的量的至少或约1-99%。如果包括第三药剂来抑制交感神经系统,则所述药剂的相对量也可以相对于第一和第二药剂变化。例如,相对于彼此,第三药剂的量可以是第一或第二药剂的量的至少或约1-99%。如果第三药剂包括在组合物中和/或用于治疗方案中,则可以允许以低于在第三药剂不存在下预期或功效所需的量使用第一和/或第二药剂。
下面进一步描述的药物组合物可以包括标准成分,如载体和防腐剂。所述组合物还可以包括增加活性成分的溶解度的物质(例如聚乙二醇)。通常,活性成分占整个组合物的少数。例如,第一、第二和/或第三药剂可以构成药物组合物的约1-50%(例如药物组合物的约1-40%;1-30%;1-20%;1-10%;2-40%;2-30%;2-20%;2-10%;2-5%;3-40%;3-30%;3-20%;3-10%;3-5%;4-40%;4-30%;4-20%;4-10%;4-5%;1-2%;1-3%;1-4%;2-4%;2-3%;或3-4%)。
当药剂“靶向”患者神经系统或内分泌系统内的区域时,它以赋予患者益处的方式影响所述区域内细胞的活动。例如,在患者对某种物质或活动成瘾的情况下,所述益处可以是减少患者使用所述物质或从事所述活动。例如,患者可以与治疗不存在下所预期相比较更不频繁或在更小程度上或与治疗之前相比在更小程度上使用所述物质或进行所述活动。因此,所述益处可以表征为复发风险降低,甚至在条件环境线索存在下。临床益处可以是主观的,正如患者可以报告他们对物质或活动的渴望减少。因此,本发明的化合物和方法可以用于促进戒断或戒断期,所述戒断期比治疗不存在下所预期的更长。实现任何可检测的改善构成用本发明的组合物和方法进行的成瘾的“治疗”;可以实现完全恢复,但构成治疗不要求完全恢复。其它适应症也是如此。例如,在焦虑相关病症、进食障碍、睡眠障碍、精神分裂症或更年期不快症状的情况下的可检测的改善构成治疗。不要求完全没有任何问题。
虽然应理解在治疗过程中发生的某些事件,但本发明的组合物不限于通过影响任何特定细胞机制起作用的那些组合物。一种假设是,就成瘾而言,触发不期望行为(例如成瘾行为)复发的线索通过条件活化影响前额叶皮质中的神经元活动的hpa轴来产生那些行为(或对行为的期望)。更具体地说,hpa轴的条件活化增加了促皮质素释放激素(crh)、促肾上腺皮质激素(acth)和皮质醇(大鼠中为皮质酮)的分泌,并且这些激素又影响前额叶皮质(大鼠中为内侧前额叶皮质)中的活动,所述前额叶皮质是参与与复发倾向有关的奖励、判断和其它活动的脑区。当施用以治疗成瘾时,本文所述的组合疗法被认为通过降低hpa轴和/或前额叶皮质内的活动来减少复发的可能性。这将线索诱导的crh、acth和/或皮质醇分泌降低到过低水平而不能引起与对物质或不期望行为的成瘾相关的渴望。随着这些激素(crh、acth和皮质醇)影响前额叶皮质中的活动,前额叶活动也可以随后下降,并且组合物的第二药剂能够促进这种下降。
如所述,本发明组合物的活性成分可以组合在单一制剂中或通过其包装组合。因此,本发明的特征在于试剂盒,所述试剂盒含有单一制剂和/或双重包装制剂以及其使用说明书。例如,组合物可以组合在单个片剂或胶囊内或者在片剂之间分开并置于泡罩包装内,任选地进行标记以指示应该服用所述组合物的日期或时刻。
如所述,所述组合物可以用于治疗对各种化合物(即治疗物质滥用)或活动的成瘾。例如,所述组合物可以用于治疗对以下各项的成瘾:兴奋剂(例如可卡因、安非他明、甲基安非他命、哌醋甲酯(methylphenidate)和相关兴奋剂)、阿片剂(例如海洛因、可待因、氢可酮和相关阿片样药物)、烟碱、酒精、处方药(例如为疼痛控制开出的药物,如
可替选地或附加性地,本文所述的组合物可以用于治疗涉及hpa轴活动和前额叶皮质的其它神经精神病症。这些病症包括焦虑,包括但不限于与恐慌症、强迫症(ocd)、创伤后应激障碍(ptsd)、社交焦虑症、广泛性焦虑症和肥胖相关的焦虑。还可以治疗被诊断为罹患抑郁的患者。他们的抑郁能够,但不是必需,与重度抑郁症、情绪不良、双相抑郁、医学病状相关抑郁以及药物滥用相关抑郁有关。
适合治疗的其它病状是肥胖和各种进食障碍,包括praderwilli综合征。适合治疗的其它患者包括罹患精神分裂症的患者;具有破坏性行为障碍(例如注意力不足障碍(add)或adhd)的患者;经历更年期的患者;以及罹患月经周期相关综合征(例如pms)的患者。适合治疗的其它病状是失眠和各种睡眠障碍。
在附图和以下描述中阐述了本发明的一个或多个实施方式的细节。本发明的其它特征、目的和优点将从说明书和附图以及权利要求书显而易见。
附图说明
图1a和1b是说明甲吡酮和奥沙西泮的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。可卡因输注的数量如图1a中所绘制,并且表示为基线的百分比的输注数量如图1b中所绘制。
图2a和2b是说明甲吡酮和奥沙西泮的组合对大鼠中静脉内自身给药三种不同剂量的可卡因的作用的柱状图。每个训练期的输注数量如图2a中所绘制,并且表示为基线的百分比的相同结果如图2b中所绘制。
图3是说明酮康唑和阿普唑仑的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。绘制了输注数量。
图4是说明酮康唑和阿普唑仑的组合对大鼠中静脉内自身给药三种不同剂量的可卡因的作用的柱状图。
图5是说明酮康唑和奥沙西泮的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。将输注表示为基线的百分比。
图6是说明cp-154,526和奥沙西泮的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。将输注表示为基线的百分比。
图7是说明甲吡酮和阿普唑仑的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。
图8是说明蝇蕈醇和cp-154,526的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。将输注表示为基线的百分比。
图9是说明蝇蕈醇和甲吡酮的组合对大鼠中静脉内可卡因自身给药的作用的柱状图。将输注表示为基线的百分比。
图10是说明甲吡酮和奥沙西泮的组合对大鼠中线索诱导的已消退可卡因寻求行为的恢复的作用的柱状图。
图11是说明长期注射甲吡酮对大鼠中线索诱导的已消退可卡因寻求行为的恢复的作用的柱状图。
图12是说明cp-154,526和奥沙西泮的组合对大鼠中线索诱导的已消退可卡因寻求行为的恢复的作用的柱状图。
图13是表示用于合成甲吡酮的合成途径的示意图。
图14是概述可卡因、甲吡酮和奥沙西泮的药代动力学分析的测试条件和结果的表格。
表1是概述作为物质滥用障碍的潜在治疗的甲吡酮和奥沙西泮的组合的安全性和药代动力学的i期单次和多次上升剂量研究的设计和人口统计资料的表格。
表2是显示作为物质滥用障碍的潜在治疗的甲吡酮和奥沙西泮的组合的安全性和药代动力学的i期单次和多次上升剂量研究的安全性(耐受性)结果的表格。
表3是显示作为物质滥用障碍的潜在治疗的甲吡酮和奥沙西泮的组合的安全性和药代动力学的i期单次和多次上升剂量研究的hpa实验室结果、病征和症状的表格。
表4是显示作为物质滥用障碍的潜在治疗的甲吡酮和奥沙西泮的组合的安全性和药代动力学的i期单次和多次上升剂量研究的药代动力学结果的表格。
具体实施方式
本文所述的组合物和方法包括两种或更多种用于治疗成瘾、其它神经精神病症以及独立或相关病状的治疗剂。制剂中包括的一种或多种药剂可以是目前可获得但目前不会为本文所述适应症开出的药剂。例如,甲吡酮通常用于诊断肾上腺功能障碍,并且奥沙西泮是用于治疗焦虑和相关疾病的苯并二氮杂卓。这两种药物影响与应激和随后的hpa轴活化有关的生理学系统。可替选地,可以根据本文的教导新形成一种或多种药剂。例如,给定所发现的靶标(let、crh、acth、gaba受体(例如gabaa或gabaa受体复合物的组分,所述组分可由本文所述的任何“第二”药剂靶向))或交感神经系统中的β肾上腺素能受体的序列,可以产生介导rnai的反义低聚核苷酸或rna分子。这些靶标的序列对于本领域普通技术人员来说是已知的或容易获得的,制备介导rnai的反义低聚核苷酸和rna分子的方法也是如此。其它有用的药剂,无论是先前可获得的还是新制备的,包括特异性结合本文所鉴定的配体(例如crh、acth或gaba)或响应于条件环境线索而被活化的受体(例如crh、acth、皮质醇或gaba的受体)的抗体。如果使用药剂来抑制交感神经系统中的活动,则所述药剂可以是化学化合物,如本文提供的化学化合物,或其它类型的药剂。例如,可以施用核酸或基于核酸的药剂来抑制β肾上腺素能受体的表达或施用特异性结合并拮抗这些受体的抗体。在特异性结合后,抗体可以用作结合的实体的激动剂或拮抗剂,根据需要促进或抑制由受体结合介导的细胞活动。例如,特异性结合crh的抗体可以用作crh拮抗剂;特异性结合gaba受体的抗体可以用作gaba受体激动剂;特异性结合β肾上腺素能受体的抗体可以用作肾上腺素拮抗剂;特异性结合糖皮质激素受体的抗体可以用作拮抗剂来抑制皮质醇;等等。
以前的实验室测试展示,hpa轴在药物成瘾中起着重要作用(goeders,psychoneuroendocrinology22:237,1997;goeders,j.pharmacol.exp.ther.301:785-789,2002;goeders,psychoneuroendocrinology27:13-33,2002;goeders,eur.neuropsychopharmacology3:435-441,2003),并且现在有数据显示某些药物组合(例如甲吡酮与奥沙西泮的组合)有效治疗成瘾(如通过减少可卡因奖励所证明)。因此,本发明的特征在于代表组合治疗剂的组合物(例如靶向本文所述的神经和/或内分泌系统区域(例如hpa轴和交感神经系统)的两种或三种药剂的组合)和用这些药剂(例如用本文所述的“第一”和“第二”药剂或“第一”和“第三”药剂)治疗患者的方法。
无论患者对何种物质或活动成瘾,成瘾的程度都可以不同;其可能在较大或较小程度上影响患者参与或应对生活中的日常事件的能力,并且可能以不同的频率复发(例如,患者可能经历极少的复发或相当规律和/或频繁的复发)。
所述药剂可以以各种方式分类,并且本发明的组合物可以包括两种或更多种相同或不同类型的药剂。例如,药剂可以归类为化学化合物(例如甲吡酮和托吡酯);蛋白质或基于蛋白质的分子,如类似抗体的突变配体(例如结合但不活化或完全活化其同源受体的配体);或核酸或基于核酸的实体,如介导rnai的反义低聚核苷酸或rna分子。因此,本发明的组合物可以包括两种或更多种化学化合物;两种或更多种不同的蛋白质或基于蛋白质的分子;或两种或更多种不同的核酸或基于核酸的实体。或者,组合物可以包括两种不同类型的药剂(例如蛋白质和核酸或化学化合物和蛋白质如抗体或其活性片段)。治疗患者的方法可以类似地包括施用两种或更多种化合物;两种或多种不同的蛋白质或基于蛋白质的分子;两种或更多种不同的核酸或基于核酸的实体;或这些各种类型的药剂的任何组合(例如,蛋白质和核酸)。
靶向hpa轴的药剂和靶向前额叶皮质的药剂中的任一者或两者可以与抑制交感神经系统中的活动的药剂组合。这些类型的药剂中的任一者或两者可以与β-阻滞剂(下文提供其合适的实例)或另一类抗高血压药和/或抗焦虑药(例如血管紧张素ii抑制剂,如坎地沙坦(candasartan))组合。第三药剂(即除了靶向hpa轴的药剂和/或靶向前额叶皮质的药剂之外的所使用的药剂)也可以是抗抑郁药,包括ssri(选择性血清素再摄取抑制剂)类中的任何药剂。
有用的化学化合物:适用于靶向hpa轴的药剂包括甲吡酮和酮康唑。甲吡酮通过抑制肾上腺皮质类固醇合成中的11β-羟基化步骤来抑制皮质酮合成(sonino,agarwal(编),hormoneantagonists,walterdegruyter,berlin中,第421-429页,1982;haleem等人,brainres.458,339-347,1988;haynes,gilman等人(编),thepharmacologicalbasisoftherapeutics,第8版,pergamonpress,newyork中,第1431-1462页,1990)。
甲吡酮可商购并且可以由合同制造商(例如,制药服务公司)合成。在一种方案中,甲吡酮可以通过两步法合成,在该方法中,将起始材料暴露于紫外光(参见例如图13中所说明的合成途径)。
可以通过测量皮质酮的血浆浓度来评定甲吡酮施用的作用。还研究了皮质酮合成抑制剂甲吡酮和酮康唑对可卡因自身给药的作用(见下文)。用甲吡酮预治疗使血浆皮质酮和正在进行的可卡因自身给药产生显著的剂量相关的减少,表明皮质酮参与可卡因奖励(还参见goeders等人,brainres.722:145-152,1996)。
酮康唑是一种广谱活性和低毒性的口服抗霉菌剂,其被用于治疗真菌病(sonino,agarwal(编),hormoneantagonists,walterdegruyter,berlin中,第421-429页,1982;thienpont等人,experientia35:606-607,1979)。这种药物还抑制肾上腺皮质类固醇合成中的11β-羟基化和18-羟基化步骤(engelhardt等人,klin.wochenschr.63:607-612,1985),并且还可以用作糖皮质激素受体拮抗体(loose等人,j.clin.invest.72:404-408,1983)。此外,临床试验表明,酮康唑(以及甲吡酮)可有效治疗对标准抗抑郁疗法有抵抗性的高皮质醇抑郁(ghadirian等人,biol.psychiatry37:369-375,1995;murphy等人,j.clin.psychopharmacol.11:121-126,1991;wolkowitz等人,am.j.psychiatry150:810-812,1993)。
抑制crh的药剂包括[met18,lys23,glu27,29,40,ala32,41,leu33,36,38]crf9-41,其缩写为α-螺旋crf(9-41)并具有序列asp-leu-thr-phe-his-leu-leu-arg-glu-met-leu-glu-met-ala-lys-ala-glu-gln-glu-ala-glu-gln-ala-ala-leu-asn-arg-leu-leu-leu-glu-glu-ala(seqidno:1),以及其生物学活性片段或变体(rivier等人,science224:889,1984)。另一种抑制crh的药剂是[d-phe12,nle21,38,(αmeleu37)]crf(12-41),其缩写为d-phecrf12-41,以及其生物学活性片段和变体。抑制crh的其它药剂包括
为了抑制acth,可以施用足量acth以通过反馈抑制来抑制acth,或下调acth受体。可以在各种分析中测试化合物影响acth的能力,所述分析包括使用例如单层培养物中的大鼠垂体前叶细胞的细胞培养物分析(参见endocrinol.91:562,1972)。
抑制hpa轴内活动的药剂还包括物质p拮抗剂(例如[d-arg1,d-pro2,d-trp7,9,leu11]sp)和血管加压素拮抗剂。
如所述,除甲吡酮、酮康唑或另一种抑制hpa轴的药剂之外,本发明的治疗剂还可以包括一种或多种通过靶向gaba来靶向前额叶皮质的药剂。苯并二氮杂卓(例如奥沙西泮)是在这方面有用的一类药物。苯并二氮杂卓属于用于药理学控制焦虑的最广泛开出的药物(baldessarini,hardman等人(编),goodman&gilman'sthepharmacologicalbasisoftherapeutics,mcgraw-hill,newyork中,第399-430页,1996)。由于与可卡因戒断相关的一些主要症状通常包括严重焦虑、烦躁不安和激动(crowley,fisher等人(编),cocaine:clinicalandbiobehavioralaspects,oxforduniversitypress,newyork中,第193-211页,1987;gawin和ellinwood,ann.rev.med.40:149-161,1989;tarr和macklin,pediatricclinicsofnorthamerica34:319-331,1987),苯并二氮杂卓可用于在戒断早期阶段减轻这些负面症状,并且并入本文所述组合疗法中的苯并二氮杂卓可以用于治疗展现这些和类似症状(即焦虑、烦躁不安和激动)的患者,无论是在成瘾情形下还是与另一个事件(例如另一个神经精神事件、更年期或pms)有关。这些药物还适用于在急诊室中治疗与可卡因中毒相关的一些医学并发症,因为急性过量后痉挛通常很明显。这些癫痫发作可以用静脉内地西泮
有用的苯并二氮杂卓或靶向前额叶皮质的药剂包括上述奥沙西泮以及利眠宁、米氮平、托莫西汀、加巴喷丁(neurontintm)、蝇蕈醇、普罗加比、利鲁唑、巴氯芬、氨己烯酸、丙戊酸(depakotetm)、塞加宾(gabitriltm)、拉莫三嗪(lamictaltm)、苯妥英(dilantintm)、卡马西平(tegretoltm)以及托吡酯(topamaxtm)。
其它有用的苯并二氮杂卓包括劳拉西泮(lorazepam)
如果包括抑制交感神经系统中的活动的药剂,则所述药剂可以是β-阻滞剂或另一类抗高血压药。更具体地,该药剂可以是索他洛尔(sotalol)
可替选地或另外地,当包括抑制交感神经系统中的活动的药剂时,所述药剂可以是ssri。目前可获得的ssri,其中任何一种或其任何组合可以用于本发明的组合物和方法,包括西酞普兰(citalopram)
靶向交感神经系统并且可归类为抗焦虑药的其它有用药剂是血管紧张素ii抑制剂,并且这些药剂包括坎地沙坦(candasartan)
苯并二氮杂卓是抗焦虑药,并且它们可以作为靶向前额叶皮质的药剂和/或作为抑制交感神经系统的药剂并入本发明的组合物中。
本发明的特征在于任何本发明化合物(即本文一般地或具体地提出的用于组合使用的任何化合物)的药学上可接受的盐、溶剂化物或水合物以及其前药、代谢物、结构类似物、多晶型物和药学上有用的其它变体,其以晶体形式、研磨和稳定为纳米晶体形式或以非结晶形式存在。这些其它变体可以是例如,含有化合物(例如甲吡酮)和如下文进一步描述的靶向部分或可检测标志物(例如,该化合物可以与荧光化合物连接或可以包含放射性同位素)的复合物。当为前药的形式时,化合物可以在施用于患者或培养中的细胞后在体内(例如细胞内)进行修饰。经修饰的化合物(即经加工的前药)可以与本文所述的化合物相同,并且具有生物学活性或具有临床上有益的足够活性。代谢物也是如此;给定化合物可以在细胞内修饰,但仍保留临床上有用的足够的生物学活性。
基于核酸的治疗剂:适用于治疗本文所述病状的治疗剂也可以是核酸。这些核酸可以通过直接或间接抑制crh、acth或皮质醇的表达而用作靶向hpa轴的第一药剂,并且其可以通过增加gaba用作靶向前额叶皮质的第二药剂。如果将第一和第二药剂中的任一者或两者与抑制交感神经系统的第三药剂组合使用,则“第三”药剂可以是抑制交感神经系统中的神经递质或其同源受体的表达的核酸(例如,核酸可以抑制β肾上腺素能受体的表达)。
核酸可以是“分离的”或“纯化的”(即不再与核酸在体内天然结合的一些或所有侧接核酸序列或细胞组分结合)。例如,相对于曾与其天然结合的细胞、组织或生物体,适用作治疗剂的核酸序列的纯度可以是至少50%(例如纯度为60%、70%、75%、80%、85%、90%、95%、98%或99%)。如果施用天然存在的或修饰的核酸序列(例如cdna),则其可包括与天然存在的基因结合的5'或3'非编码序列中的一些。例如,分离的核酸(dna或rna)可以包括侧接编码序列的5'或3'非编码序列中的一些或全部(例如,转录成mrna中的启动子或增强子的dna序列,或产生mrna中的启动子或增强子的rna序列)。例如,分离的核酸可以含有小于约5kb(例如,小于约4kb、3kb、2kb、1kb、0.5kb或0.1kb)的5'和/或3'序列,所述序列在天然存在所述核酸的细胞中天然侧接于所述核酸分子。倘若核酸是rna或mrna,则当它基本上不含在细胞中与其天然结合的细胞组分以及在细胞进行培养的情况下基本上不含细胞组分和培养细胞的培养基时(例如当rna或mrna为含有少于约20%、10%、5%、1%或更少其它细胞组分或培养基的形式时),则它是从天然来源(例如组织)或细胞培养物中“分离的”或“纯化的”。在化学合成时,在核酸基本上不含其合成中使用的化学前体或其它化学物质时(例如,当核酸为含有少于约20%、10%、5%、1%或更少化学前体或其它化学物质的形式),核酸(dna或rna)是“分离的”或“纯化的”。
适用于本文所述的组合物和方法的核酸可以是双链或单链的,并且因此可以是有义链、反义链或者有义或反义链的一部分(即片段)。可以使用标准核苷酸或核苷酸类似物或衍生物(例如肌苷、硫代磷酸酯或吖啶取代的核苷酸)合成核酸,所述核苷酸类似物或衍生物可以改变核酸与互补序列配对或抵抗核酸酶的能力。可以通过修饰核酸的碱基部分、糖部分或磷酸骨架来改变(例如改善)核酸的稳定性或溶解性。例如,本发明的核酸可以如toulme(naturebiotech.19:17,2001)或faria等人(naturebiotech.19:40-44,2001)所教导来修饰,并且核酸的脱氧核糖磷酸骨架可以被修饰来产生肽核酸(pna;参见hyrup等人,bioorganic&medicinalchemistry4:5-23,1996)。
pna是核酸“模拟物”;分子的天然骨架被假肽骨架取代,并且只保留了四个核苷酸碱基。这允许在低离子强度的条件下与dna和rna特异性杂交。pna可以使用标准固相肽合成方案合成,如例如hyrup等人(同上)和perry-o'keefe等人(proc.natl.acad.sci.usa93:14670-675)所述。本文所述的核酸的pna可以用于治疗和诊断应用中。例如,pna可用作反义或反基因试剂来通过例如诱导转录或翻译停滞或抑制复制进行基因表达的序列特异性调节。
核酸可以在施用于患者之前被并入载体(例如自主复制的质粒或病毒)中,并且这些载体在本发明的范畴内。本发明还涵盖以有义或反义方向包括本发明核酸的遗传构建体(例如质粒、粘粒和转运核酸的其它载体)。核酸可以与促进核酸表达的调节序列(例如启动子、增强子或其它表达控制序列,如多腺苷酸化信号)可操作地连接。载体可以自主复制或整合到宿主基因组中,并且可以是病毒载体,如复制缺陷型逆转录病毒、腺病毒或腺相关病毒。另外,当存在时,调节序列可以指导核酸的组成型或组织特异性表达。
核酸可以是反义低聚核苷酸。在与靶序列的编码链“反义”时,它们不需要与编码序列结合;它们还可以与非编码区(例如5'或3'非翻译区)结合。例如,反义低聚核苷酸可以与mrna的翻译起始位点周围的区域互补(例如在目的靶基因的-10和+10区域之间或在多腺苷酸化信号中或周围)。此外,可以通过靶向与调节区(例如启动子和/或增强子)互补的核苷酸序列以形成阻止靶细胞中基因转录的三螺旋结构来抑制基因表达(一般地参见helene,anticancerbioassaysdrugdes.6:569-84,1991;helene,ann.n.y.acad.sci.660:27-36,1992;和maher,14:807-15,1992)。可以通过产生所谓的“回转”核酸来增加能够以这种方式成功靶向的序列。回转分子:以交替的5'-3'、3'-5'方式合成,使得它们与双链体的第一链碱基配对,然后与另一个链碱基配对,从而无需在双链体的一条链上具有嘌呤或嘧啶的大片段。
具有少至9-10个核苷酸(例如12-14、15-17、18-20、21-23或24-27个核苷酸;sirna通常具有21个核苷酸)的片段可以是有用的并且属于本发明的范畴。
在其它实施方式中,反义核酸可以是异头核酸,其与互补rna形成特异性双链杂交体,在所述杂交体中,与通常的b-单元相反,链彼此平行延伸(gaultier等人,nucleicacidsres.15:6625-6641,1987;还参见tanaka等人,nucl.acidsres.22:3069-3074,1994)。或者,反义核酸可以包含2'-邻甲基核糖核苷酸(inoue等人,nucleicacidsres.15:6131-6148,1987)或嵌合rna-dna类似物(inoue等人,fesslett.215:327-330,1987)。
抗体:适用作本发明组合物中的治疗剂的抗体和其抗原结合片段。这些抗体可以是g类(igg),但也可以使用igm、igd、iga和ige抗体;需要的是,抗体以根据一些发现赋予其所施用的患者临床利益的方式特异性结合本文所述的靶标并且通过提高或者抑制靶标活性来改变该靶标(。抗体可以是多克隆或单克隆抗体,并且术语“抗体”用于指整个抗体或其片段吗,所述片段是或包括整个抗体的抗原结合域。例如,有用的抗体可能缺乏fc部分;可以是单链抗体;或可以是由抗体的可变抗原结合域组成(或基本上由其组成)的片段。所述抗体可以是人源化的(通过例如cdr移植)或完全人类的。
产生抗体的方法是本领域中众所周知的。例如,如上所述,可以在携带人类免疫球蛋白基因而不是小鼠免疫球蛋白基因的转基因小鼠中产生人类单克隆抗体。可以使用从这些小鼠(用目的抗原进行免疫后)获得的脾细胞产生分泌人类mab的杂交瘤,所述人类mab对来自人类蛋白质的表位具有特异性亲和力(参见例如wo91/00906、wo91/10741;wo92/03918;wo92/03917;lonberg等人,nature368:856-859,1994;green等人,naturegenet.7:13-21,1994;morrison等人proc.natl.acad.sci.usa81:6851-6855,1994;bruggeman等人,immunol.7:33-40,1993;tuaillon等人,proc.natl.acad.sci.usa90:3720-3724,1993;和bruggeman等人,eur.j.immunol21:1323-1326,1991)。
抗体也可以是可变区或其部分(例如cdr)在非人类生物体(例如大鼠或小鼠)中产生的抗体。因此,本发明涵盖嵌合的、cdr-移植的和人源化的抗体,其在非人类生物体中产生,接着被修饰(在例如可变骨架或恒定区中)来降低在人体内的抗原性。嵌合抗体(即不同部分来源于不同动物物种的抗体(例如,鼠mab的可变区和人类免疫球蛋白的恒定区))可以通过本领域中已知的重组技术产生。例如,可以用限制酶消化编码鼠(或其它物种)单克隆抗体分子的fc恒定区的基因来去除编码鼠fc的区域,并且用编码人类fc恒定区的基因的等效部分来代替(参见欧洲专利申请号125,023;184,187;171,496;和173,494;还参见wo86/01533;美国专利号4,816,567;better等人,science240:1041-1043,1988;liu等人,proc.natl.acad.sci.usa84:3439-3443,1987;liu等人,j.immunol.139:3521-3526,1987;sun等人,proc.natl.acad.sci.usa84:214-218,1987;nishimura等人,cancerres.47:999-1005,1987;wood等人,nature314:446-449,1985;shaw等人,j.natl.cancerinst.80:1553-1559,1988;morrison等人,proc.natl.acad.sci.usa81:6851,1984;neuberger等人,nature312:604,1984;和takeda等人,nature314:452,1984)。
本发明的抗原结合片段可以是:(i)fab片段(即由vl、vh、cl和ch1域组成的单价片段);(ii)f(ab')2片段(即含有在铰链区通过二硫键连接的两个fab片段的二价片段);(iii)由vh和ch1域组成的fd片段;(iv)由抗体单臂的vl和vh域组成的fv片段,(v)dab片段(ward等人,nature341:544-546,1989),其由vh域组成;以及(vi)分离的互补决定区(cdr)。
可以使用表达载体离体(例如本发明的蛋白质可以从表达系统如本文所述的表达系统纯化)或体内(在例如整个生物体中)产生本发明的蛋白质,包括抗体。
制剂和剂量:靶向hpa轴、前额叶皮质和/或交感神经系统的鉴定的药剂可以以治疗有效剂量施用于患者来预防、治疗或改善本文所述的任何病症或病状(例如成瘾、肥胖、创伤后应激障碍或相关病状)。治疗有效剂量是指足以改善病症或病状的至少一种病征或症状的药剂或药剂组合的量。
适用于本发明情形的许多药剂先前已出于其它理由用于治疗患者。如果可获得剂量信息,则其可用于帮助确定药剂在当前描述的组合中的有效剂量。用于治疗患者成瘾——本文所述的其它疾病之一——和/或相关病状的剂量可以与先前用于另一种适应症的剂量相同。所述剂量也可以不同。例如,与本文所述的组合疗法相关的所需有效剂量可以小于先前被证明安全且有效的剂量。
如果需要,可以通过细胞培养物或实验动物中的标准药学程序确定本文所述药剂的毒性和治疗功效。例如,可以使用实验动物如啮齿动物和非人灵长类动物来测定ld50(对50%群体致死的剂量)和ed50(在50%群体中治疗有效的剂量)。毒性和治疗效果之间的剂量比是治疗指数,其可以表示为比率ld50:ed50。展现大治疗指数的化合物通常是优选的。
从细胞培养物分析和动物研究获得的数据可以用于配制用于人类的一系列剂量。这些化合物的剂量优选落在包括ed50的循环浓度范围内,几乎没有毒性。剂量可以根据所用的剂型和所用的施用途径在这个范围内变化。对于本发明方法中使用的任何化合物,治疗有效剂量可以首先从细胞培养物分析(例如设计来判定核酸、基于核酸的药剂或蛋白质如抗体是否抑制(或刺激)其旨在抑制(或刺激)的配体或受体的表达或活性的分析)估计。
可以在动物模型中配制剂量来实现包括细胞培养物中所测定的ic50(即测试化合物实现症状的半数最大抑制的浓度)的循环血浆浓度范围。这些信息可以用于更准确地确定在人体中的有用剂量(例如治疗有效剂量)。例如,可以通过高效液相色谱法测量血浆中的水平。
治疗药物成瘾的最大问题之一是再犯率高。这种现象可以在作为广泛视为药物摄取复发倾向的临床前模型的在恢复期间的动物中测试,并且可以使用恢复动物模型进一步确定和限定本文所述的药剂的有效剂量。例如,可以教导动物自身给药药物直到维持稳定的药物摄入,然后进行长期消退训练或戒除。满足消退标准后或在规定的戒除期后,特定刺激恢复对先前与药物输注物递送相关的操作进行反应的能力被视为寻求药物的量度。这种药物寻求行为的恢复可以通过在大鼠和猴子中引发注射药物自身(stewart,j.psychiatr.neurosci.25:125-136,2000)或通过使大鼠暴露于短时间间歇性足电击(shaham等人,brainres.rev.33:13-33,2000;stewart,j.psychiatr.neurosci.25:125-136,2000)引发。急性再暴露于自身给药的药物(dewit,exp.clin.psychopharmacol.4:5-10,1996)和暴露于应激(shiffman和wills,copingandsubstanceabuse,academicpress,orlando,1985;lamon和alonzo,addict.behav.22:195-205,1997;brady和sonne,alc.res.health23:263-271,1999;sinha,psychopharmacol.158:343-359,2001;和sinha等人,psychopharmacol.142:343-351,1999)或简单地呈现应激相关的图像(sinha等人,psychopharmacol.158:343-359,2000)也被鉴定为是引起人类药物寻求复发的有效事件。
在下述研究中,最初找到甲吡酮和奥沙西泮中的每一种减少可卡因自身给药而不会对其它行为产生非特异性削弱作用的剂量。然后将该剂量减少一半,直到找出每种药物不再影响可卡因自身给药或任何其它可观察到的行为的剂量(即无效剂量)。接着组合两种药物的无效剂量,可卡因自身给药减少。这表明尽管这两种药物通过不同的机制产生其作用,但所述作用是加和性的。因此,似乎将通过不同机制影响hpa轴的药物进行组合可以对可卡因奖励产生加和或协同作用。此外,通过将这些药物以所述药物单独施用时无效的浓度组合,可以将可能与这些化合物相关的潜在毒副作用(例如,甲吡酮引起的血浆皮质醇过量降低以及苯并二氮杂卓的滥用倾向)降至最低。因此,本发明的组合物可以包括治疗剂的组合,其中一种或两种的剂量水平低于所述药剂单独施用时实现作用所需的剂量水平;所述剂量可以是加和性的。
可以使用一种或多种生理学上可接受的载体或赋形剂以任何常规方式配制根据本发明使用的药物组合物。因此,可以配制药剂,包括化合物和其生理学上可接受的盐和溶剂化物,用于通过口服或肠胃外施用进行施用。
为进行口服施用,药物组合物可以采取例如通过常规方法用药学上可接受的赋形剂制备的片剂或胶囊的形式,所述赋形剂如粘合剂(例如预胶化玉米淀粉、聚乙烯吡咯烷酮或羟丙基甲基纤维素);填充剂(例如乳糖、微晶纤维素或磷酸氢钙);润滑剂(例如硬脂酸镁、滑石或二氧化硅);崩解剂(例如马铃薯淀粉或淀粉羟基乙酸钠);或润湿剂(例如十二烷基硫酸钠)。片剂可以通过本领域中众所周知的方法包被。用于口服施用的液体制剂可以采取例如溶液、糖浆或悬浮液的形式,或其可以以干燥产品的形式呈递以在使用前用水或其它合适的载体构造。这些液体制剂可以通过常规方法利用药学上可接受的添加剂制备,所述添加剂如悬浮剂(例如山梨糖醇糖浆、纤维素衍生物或氢化食用脂肪);乳化剂(例如卵磷脂或阿拉伯胶);非水性载体(例如杏仁油、油性酯、乙醇或分级植物油);以及防腐剂(例如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯或山梨酸)。制剂还可以适当含有缓冲盐、调味剂、着色剂和甜味剂。
可以合适地配制用于口服施用的制剂以提供活性化合物(其在本文中可称为“治疗剂”)的控制释放。
可以配制包括化合物(例如小有机分子)的药剂以通过注射(例如通过弹丸式注射或连续输注)进行肠胃外施用。用于注射的制剂可以以单位剂型(例如,在安瓿或多剂量容器中)与添加的防腐剂一起提供。组合物可以采取如油性或水性载体中的悬浮液、溶液或乳液的形式,并且可以含有配制剂,如悬浮剂、稳定剂和/或分散剂。或者,活性成分可以为粉末形式以在使用前用合适的载体(例如无菌无热原水)构造。
除了先前描述的制剂之外,还可以将药剂配制成储库制剂。这种长效制剂可以通过植入(例如皮下或肌肉内)或通过肌肉内注射施用。因此,例如,所述药剂可以用合适的聚合或疏水材料(例如作为可接受的油中的乳液)或离子交换树脂配制,或配制成微溶衍生物,例如微溶盐。
所述组合物还可以被配制来用于其它施用途径,包括鼻内、局部和粘膜(例如通过舌下施用)。
如果需要,组合物可以呈递在可含有一个或多个含有活性成分的单位剂型的包装或分配器装置中。所述包装可以例如包含金属或塑料箔,如泡罩包装。包装或分配器装置可以附有关于施用的说明书。各种呈递形式(例如通过包装和分配器呈递)都属于本发明的范畴。
核酸,包括反义核酸,也可以全身施用,并且如果进行全身施用,则可以进行修饰来靶向hpa轴、前额叶皮质和/或交感神经系统内的所选细胞。例如,反义核酸可以与抗体或其它蛋白质(例如受体配体)连接,所述抗体或其它蛋白质特异性结合细胞表面受体或与靶细胞类型相关的其它组分。类似地,核酸可以包括促进其转运跨过细胞膜(参见例如letsinger等人,proc.natl.acad.sci.usa86:6553-6556,1989;lemaitre等人,proc.natl.acad.sci.usa84:648-652,1987;和wo88/09810)或血脑屏障(参见例如wo89/10134)的药剂。此外,核酸可以用嵌入剂修饰(zon,pharm.res.5:539-549;1988)。还可以使用本文描述的载体将反义核酸递送至细胞。为了获得足够的细胞内反义核酸浓度,可以在具有强启动子的载体(例如,强polii或poliii启动子)中表达所述反义核酸。
在特定实施方式中,本发明的特征在于药物组合物,所述药物组合物包括靶向hpa轴的第一药剂和靶向前额叶皮层的第二药剂。第一药剂可以是抑制crh、抑制acth和/或抑制皮质醇的药剂,并且第二药剂可以是增加gaba的表达、分泌或活性,作为gaba模拟物和/或抑制gaba代谢的药剂。第一和/或第二药剂可以是化学化合物。例如,第一药剂可以是甲吡酮
适用于运输、储存和后续稀释的浓缩组合物也属于本发明。
上述药物组合物可以用于本文所述的方法,包括以下方法,并用于下文所述的用途(例如用于制备药物和/或用于制备用于治疗本文所述病症或病状的药物)。
治疗方法:如所述,可以使用本文所述的组合物治疗罹患与hpa轴中的异常活动相关的病症的患者。治疗方法可以包括各种步骤,其中一个步骤可构成鉴定需要治疗的患者。医生能够很好地检查和诊断怀疑患有成瘾和/或本文所述的另一种病状的患者。在可在替选性方案中进行的诊断之后,医生可以开出治疗有效量的组合物(例如包含靶向hpa轴的第一药剂和靶向前额叶皮质的第二药剂的药物组合物)。患者可以患有或被诊断为患有对物质成瘾,所述物质如酒精、化学刺激剂、处方(或开出的)止痛药或天然存在的源自植物的药物。化学刺激剂可以是可卡因、安非他明、甲基安非他命或结晶甲基安非他命盐酸盐或哌醋甲酯。如果特定药物的类似物具有成瘾性,则也可以治疗对这些类似物的成瘾。
所述药物还可以是巴比妥酸盐(例如硫胺素(thiamyl)
天然存在的源自植物的药物包括大麻和烟草。可以使用本文所述的组合物治疗通常对这些物质成瘾和/或对其中更具体的成分(例如烟草中的烟碱)成瘾的患者。成瘾也可能表现为对活动如赌博、性或性活动或暴饮暴食(其可能与进食障碍相关或可能导致肥胖)成瘾。更一般地,进食和睡眠障碍属于适合用本发明组合物治疗的病症。进食障碍包括神经性厌食、神经性贪食、暴食症和未另外规定的进食障碍(ednos)。数种研究已经检查了hpa轴在神经性厌食中的功能。主要发现皮质醇增多,这与增加的中枢促肾上腺皮质激素释放激素水平和促肾上腺皮质激素的正常循环水平相关。虽然神经性厌食能够难以诊断,但患有这种病症的患者存在内分泌功能障碍,通常表现为闭经、异常的温度调节、异常的生长激素水平和异常进食。本发明方法可以包括对需要治疗的患者进行鉴定的步骤,并且这些特征会是或者可能会属于医生用于诊断神经性厌食的特征。
可以使用本发明的组合物治疗患有praderwilli综合征的患者,并且治疗这些患者的方法属于本发明的范畴。
睡眠障碍包括失眠、睡眠呼吸暂停睡眠障碍、不安腿综合征(rls)和周期性肢体运动障碍(plmd)以及发作性睡病。
适合治疗的其它患者包括罹患焦虑(其可以与恐慌症、强迫症(ocd)、创伤后应激障碍(ptsd)、社交焦虑症相关,或可以是广泛性焦虑症)的患者。如果病状是抑郁,则其可以是与重度抑郁症或情绪不良、双相抑郁相关的抑郁,或者可以与医学病状或物质滥用相关。产生抑郁或其它重度情感障碍的风险取决于遗传易感性、环境暴露和衰老之间复杂相互作用。
适合治疗的其它患者包括罹患精神分裂症的患者;患有注意力不足障碍(例如add或adhd)的患者;经历更年期的患者;以及罹患月经周期相关综合征(例如pms)的患者。
本文描述的病症和事件可以进行各种分类,并且可以以各种方式彼此相关。例如,社交焦虑可以导致进食障碍,并且其它焦虑相关病状如ptds可以表现为睡眠障碍。被诊断为临床抑郁的患者也可以经历睡眠障碍。已经表征为进行性病症的成瘾可以从自身给药处方药或非处方药来减轻另一种神经精神病症的症状开始。例如,患者可以在抑郁或焦虑的情况下自身给药酒精或大麻或自身给药睡眠助剂来治疗由抑郁或焦虑导致的睡眠困难。病症和相关病状或症状之间的关系也可以沿不同方向流动。例如,失眠中hpa轴的慢性活化使得失眠患者不仅具有精神障碍(即慢性焦虑和抑郁)的风险,而且还具有与这种活化相关的显著医学发病的风险。到目前为止,失眠是医疗实践中最常见的睡眠障碍。无论是作为各种精神或医学病症的症状还是作为应激情况的结果,慢性和严重失眠都被患者视为一种独特的病症(参见vgontzas等人,j.clin.endocrinol.metabl.86:3787-3794,2006)。睡眠障碍,包括失眠,可以发生在更年期期间或当患者罹患pms时。
正如本文所述的适应症的分类中可以存在一些重叠一样,所施用的药剂的性质和/或将其分类的方式可以存在一些重叠。例如并且如上所述,苯并二氮杂卓可用作靶向前额叶皮质的“第二”药剂。苯并二氮杂卓也可以归类为抗焦虑药物,因此适合作为本文所述的“第三”药剂。
可以通过多种方式评估治疗的成功,包括客观测量结果(例如,在患者对物质或活动成瘾的情况下,药物自身给药或其它成瘾活动的频率或严重程度降低),健康的总体改善(例如血压、肾功能、肝功能或血细胞计数的改善)和/或主观测量结果(例如患者报导对物质或活动的渴望减少或更好的健康感(例如在患者罹患焦虑或焦虑相关病症的情况下,报导焦虑减少、情绪改善、健康感增加或应对日常应激源的能力改善))。如果所治疗的病状是进食障碍或睡眠障碍,则可以通过判断正常进食或睡眠模式的有效恢复(或正朝着正常进食或睡眠模式恢复)来评估治疗。
在特定实施方式中,本发明的特征在于治疗罹患与hpa轴中的异常活动相关的病症的患者的方法。所述方法可以包括以下步骤:(a)鉴定需要治疗的患者;(b)向患者施用治疗有效量的本文所述组合物。所述病症可以包括成瘾、焦虑、精神分裂症或抑郁;所述病症可以是对物质(例如化学刺激剂,如阿片剂(如海洛因、可待因、氢可酮或其类似物)、烟碱、酒精、处方止痛药或天然存在的源自植物的药物如烟碱)成瘾。化学刺激剂还可以是可卡因、安非他明、甲基安非他命、哌醋甲酯或其类似物。这种病症也可以是对活动如赌博或从事性活动或过度进食成瘾。
如果患者罹患焦虑,则焦虑可以与恐慌症、强迫症(ocd)、创伤后应激障碍(ptsd)、社交焦虑症或广泛性焦虑症相关。如果患者罹患抑郁,则所述抑郁能够与重度抑郁症或情绪不良相关,与双相抑郁或医学病状或物质滥用相关。如所述,所述病症还可以是进食障碍或睡眠障碍或破坏性行为障碍。
可以通过以下方式执行所述方法来治疗罹患更年期或月经周期的不良症状的患者:(a)鉴定需要治疗的患者;和(b)向患者施用治疗有效量的本文所述组合物。递送的组合物的量是在治疗上是有效的,通过症状缓解来判断有效性,所述症状可以包括焦虑、抑郁或睡眠困难。
本发明的特征在于本文所述组合物在制备药物中的用途。与上下文无关,本发明的特征还在于本文所述组合物在制备用于治疗以下病症的药物中的用途:肥胖;进食障碍;睡眠障碍;抑郁;破坏性行为障碍;精神分裂症;和/或焦虑。
实施例
实施例1
低剂量组合药物疗法对大鼠可卡因自身给药的作用:这里描述的研究检查了与本文所述一致的用于治疗成瘾(更具体地,可卡因滥用)的组合药物疗法。使用这种方法,以单独时无效或有效性低得多的剂量一起施用认为使用不同的作用机制最终对身体对应激源的反应产生类似作用的两种化合物。在多个交替的可卡因和食物自身给药方案下训练成年雄性wistar大鼠。这个方案由交替周期的可卡因获取和食物强化组成。在一些情况下,如下文进一步描述,测试了可卡因的三种剂量(每次输注0.125、0.25或0.50mg/kg)。在同一训练期期间,还定期用盐水替代(可卡因消退)和食物消退训练大鼠。
这些研究支持以下结论:用皮质酮合成抑制剂甲吡酮和酮康唑、苯并二氮杂卓利眠宁、阿普唑仑和奥沙西泮以及crh受体拮抗剂cp-154,526预处理均减少了大鼠的可卡因自身给药和已消退可卡因寻求的恢复。组合药物疗法可能通过减弱线索诱导的hpa轴内活动增加,从而减少线索诱导的crh、acth和皮质醇(皮质酮)分泌以及通过减少线索诱导的前额叶皮质中活动的变化来减少复发的可能性。
测试的组合:我们测试的药物组合包括:
(1)甲吡酮和奥沙西泮;(2)酮康唑和阿普唑仑;(3)酮康唑和奥沙西泮;(4)甲吡酮和阿普唑仑;(5)蝇蕈醇和cp-154,526;以及(6)蝇蕈醇和甲吡酮。药物组合由来自每个类别的至少一种药物(例如甲吡酮和奥沙西泮)组成。如所述,药物以低于其正常有效剂量的剂量组合,并且出现加和或协同作用。
自身给药可卡因的训练:在一个模型中,使大鼠暴露于交替的15分钟周期的可卡因自身给药接口和食物强化。使用食物来控制药物和组合的潜在非特异性共济失调作用。理想的药物或药物组合是减少可卡因自身给药而不影响食物维持反应的药物或药物组合。已经使用的另一种临床前模型是线索诱导的已消退可卡因寻求的恢复复发模型。在这个模型中,训练大鼠自身给药可卡因,并评定环境中条件线索恢复已消退反应的能力并将其用作复发的量度。
更具体地,向成年雄性wistar大鼠植入长期颈静脉导管。从手术恢复后,在食物强化和可卡因自身给药的多重交替方案下训练大鼠进行反应。使用食物维持反应来控制各种治疗的非特异性运动作用。在该方案的食物分量期间,将位于食物反应杆上方的刺激光照亮来指示可获得食物强化。最初,食物反应杆的每次下压导致食物刺激光短暂变暗(0.6秒)并递送食物颗粒(45mg)。递送每个食物颗粒之后,暂停25秒。在此暂停期间,将刺激光变暗,计数对食物杆的反应,但没有预定结果。在食物分量期间对另一个(可卡因)杆的反应也没有预定结果。食物杆的反应要求经几个训练期逐渐增加,从连续强化到固定比率四次方案,其中食物呈递需要四次反应。在获取食物15分钟后,使室内的所有刺激光都变暗,暂停1分钟。暂停后,将可卡因反应杆上方的刺激光照亮来指示可获得可卡因(每次输注0.125、0.25或0.5mg/kg)。最初,可卡因反应杆的每次下压导致刺激光短暂变暗并输注可卡因(在5.6秒内递送200μl)。每次输注后都有20秒的暂停时段。对可卡因的反应要求逐渐增加到固定比率四次强化方案。在获取可卡因15分钟和暂停1分钟后,再次允许大鼠获取方案的食物分量15分钟。在两小时行为训练期期间,以这种方式每15分钟交替获取食物和可卡因,以使得每只大鼠各自暴露于食物和可卡因4个15分钟时段。每个行为训练期开始于获取食物或可卡因15分钟,并且这个操作每天交替进行。当可卡因和食物呈递的总数以及每个训练期四次暴露中的每一次期间呈递的次数在三个连续训练期中的变化少于10%时,就建立了稳定的反应基线。测试了可卡因的至少三种不同剂量(例如每次输注0.125、0.25或0.50mg/kg)。首先训练大鼠来自身给药每次输注0.25mg/kg,这是所用可卡因的标准剂量。当反应稳定时,适当地将剂量改变为每次输注0.125或0.5mg/kg。最初发现用这种中等剂量的可卡因(即每次输注0.25mg/kg)训练大鼠加快了使用较低剂量(即每次输注0.125mg/kg/)的稳定性。
获得稳定的反应基线后,为每只大鼠单独产生各种化合物的剂量-反应曲线。用每种剂量处理大鼠至少两次,其中每次测试之间间隔最少两天的基线可卡因自身给药。仅用两种测试化合物测试每组大鼠来使潜在的遗留作用达最小。确定每种化合物将可卡因自身给药减少至少50%而不影响食物维持反应的最低有效剂量(即高剂量)。选择用于药物组合实验的剂量是最低有效剂量的一半,并且该剂量还必须使可卡因自身给药降低小于10%(即,无效剂量)。如果最低有效剂量的一半使可卡因自身给药减少超过10%,则将该剂量再次减少一半。例如,酮康唑的最低有效剂量为25mg/kg,并且在阿普唑仑和奥沙西泮的研究中成功使用12.5mg/kg的剂量。在单独测试时,这个剂量(12.5mg/kg)对可卡因或食物维持反应没有作用,但当与类似无效剂量的阿普唑仑(即1.0mg/kg,腹膜内)或奥沙西泮(10mg/kg,腹膜内)组合时显著减少了可卡因自身给药。这个基本原理指导组合研究中每种化合物的剂量选择。每个实验组由3至10只大鼠组成。
线索诱导的已消退可卡因寻求的恢复:本文所述的实验被设计来研究被鉴定为有效减少可卡因自身给药的药物组合是否还会阻断条件线索恢复已消退可卡因寻求行为的能力。向成年雄性wistar大鼠植入慢性颈静脉导管,并通过在每周进行5天的每天2小时训练期期间以固定比率四次(fr4)强化方案按压实验腔室中的反应杆之一(即“有效”或“可卡因”杆)来训练自身给药可卡因(每次输注0.25mg/kg)。在每个训练期开始时,将两个杆伸入腔室中,并且将有效杆(acitvelever)上方的刺激光照亮来指示可获得可卡因。最初,有效杆的每次下压引起可卡因的静脉内输注并且同时提供房屋光和调谐复合(tonecompound)刺激(即条件线索或第二强化物)。每次输注后都有20秒的暂停时段。在暂停时段期间,将有效杆上方的刺激光以及房屋光和调谐复合刺激取消,并且一旦暂停结束就将有效杆上方的光照亮。当对有效杆的反应连续两天变化小于20%时,则将反应要求增加到fr2。当在fr2方案或强化下观察到类似的稳定性时,将反应要求增加到最终比率4。在fr4强化方案下稳定反应的标准是至少10天暴露于这个方案,当至少连续三天的反应变化小于10%时,这个方案结束。计数对无效杆(inactivelever)的反应,但在任何时候都不会产生任何程序化结果。一旦观察到稳定的可卡因自身给药后,就将大鼠暴露于消退的环境;将大鼠放入行为腔室,但对“可卡因”(有效)杆的反应不产生程序化结果。持续进行消退训练直到反应降低至基线自身给药的不到20%。接着建议进行恢复测试。将大鼠放入实验腔室中,将两个反应杆伸入腔室中,并且在自身给药训练期间将“有效”杆上方的刺激光照亮。在恢复期间,对“有效”杆的反应引起条件强化物(即在自身给药期间与可卡因配对的房屋光和调谐复合刺激)的5.6秒呈递。计数对“无效”杆的反应,但不产生任何预定结果。将在恢复测试期间对“有效”杆的反应视为可卡因寻求行为的指标。每个实验组由0.8至10只大鼠组成。
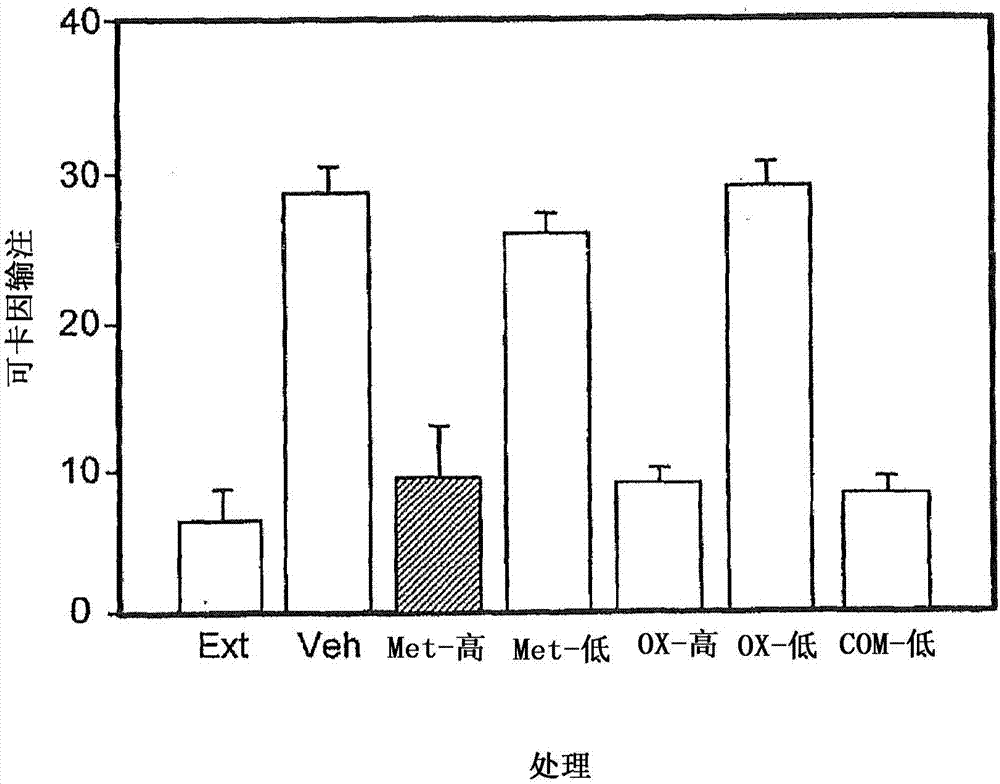
甲吡酮和奥沙西泮对静脉内自身给药可卡因的作用:这些实验被设计来确定甲吡酮和奥沙西泮的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。结果如图1a的图所描绘。左侧的第一个柱(“ext”)显示当对“有效”杆反应仅引起盐水输注时的消退结果。第二个柱(“veh”)描绘了用治疗药物的载体(0.9%盐水中的5%乳化剂)预处理后自身给药的可卡因输注的数量。“met-高”柱显示用高剂量甲吡酮(25-175mg/kg,腹膜内)预处理后可卡因输注的数量,而“ox-高”柱描绘了用高剂量的奥沙西泮(5-80mg/kg,腹膜内)预处理后自身给药的可卡因输注的数量。
这些剂量下的甲吡酮和奥沙西泮都减少了可卡因自身给药,而不会影响食物维持反应。“met-低”和“ox-低”柱表示单独用无效低剂量(奥沙西泮5-25mg/kg,腹膜内;甲吡酮25-50mg/kg,腹膜内)的甲吡酮和奥沙西泮预处理后的反应。显然,当单独施用时,这些剂量不会显著影响可卡因自身给药(或食物维持反应)。“com-低”柱描绘了在递送组合药物疗法(即由无效剂量的甲吡酮和奥沙西泮组成的注射剂)后自身给药的可卡因输注的数量。可以看出,由甲吡酮和奥沙西泮组成的组合药物疗法将可卡因自身给药减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到消退水平而不影响食物维持反应,表明该组合减少了寻求可卡因的动机而不影响另一种强化物(即食物)的反应或动机。
图1b描绘了与图1a中所示相同的数据,但数据以在测试条件下基线输注的百分比的形式呈现。“高”剂量的甲吡酮和奥沙西泮将可卡因自身给药减少到低于基线自身给药的50%,而“低”剂量仅使自身给药减少10%或更少。如图1a中所示,低剂量的奥沙西泮和甲吡酮的组合将可卡因自身给药减少到消退期间所见的水平。
图2a描绘了被设计来研究当训练不同组的大鼠自身给药不同剂量的可卡因时无效剂量的甲吡酮和奥沙西泮的组合对可卡因自身给药的作用。判定当可获得更高剂量的可卡因时大鼠是否能够克服所述组合的作用是重要的。这类似于可卡因成瘾者增加他或她的可卡因摄入来克服组合药物疗法的作用。x轴上的数字表示自身给药的可卡因的三种剂量。“盐水”显示当在注射器中只有盐水(即消退)时自身给药的输注的数量。“载体”表示在可卡因自身给药训练期开始之前递送用于药物治疗的载体(在0.9%盐水中的5%乳化剂)时自身给药的可卡因输注的数量。“组合”描述了在用无效剂量的甲吡酮和奥沙西泮的组合预处理后自身给药的可卡因输注的数量。显然,无论可用于自身给药的可卡因剂量如何,这种组合都将可卡因自身给药减少到消退水平。这指示组合药物疗法的作用不会因为增加可卡因的摄入或剂量而被容易地克服。
图2b描绘了与图2a中所示相同的数据,但数据以在不同条件下基线输注的百分比的形式呈现。图2a显示低剂量的奥沙西泮和甲吡酮的组合将可卡因自身给药减少到消退期间所见的水平,而与可用于自身给药的可卡因的剂量无关。
还进行了实验来确定甲吡酮和奥沙西泮的组合对大鼠中线索诱导的已消退可卡因寻求的恢复的作用。使用可卡因寻求的复发动物模型。参考图10,标记为“sa”的柱描绘了在可卡因自身给药期间对“有效”杆反应的次数。标记为“ext”的柱描绘了在消退期间当对“有效”杆反应仅引起盐水输注时对“有效”杆的反应次数。第三个柱“veh”表示在恢复测试期间在用治疗药物的载体(在0.9%盐水中的5%乳化剂)预处理后对“有效”杆的反应。最后一个柱“组合”描述了在恢复测试期间在递送组合药物疗法(即由无效剂量的甲吡酮和奥沙西泮组成的注射剂,如在可卡因自身给药实验中所确定(参见图1a))后对“有效”杆反应的次数。所述组合药物疗法将可卡因寻求(即在恢复期间对有效杆的反应)减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合疗法将恢复(复发)减少到消退水平,而不影响食物维持反应。这表明所述组合降低了寻求可卡因的动机而不影响对另一种强化物(即食物)的反应或动机。
酮康唑和阿普唑仑对静脉内自身给药可卡因的作用:这些实验被设计来确定酮康唑和阿普唑仑的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。数据如图3所呈现。实心柱(“veh”)描绘了用载体(0.9%盐水中的5%乳化剂)预处理后自身给药的可卡因输注的数量。空心柱(“ext”)显示当对“有效”杆反应仅引起盐水输注时的消退结果。“alp”(条纹)和“keto”(不紧密阴影)柱表示单独用无效低剂量(阿普唑仑0.2-2mg/kg,腹膜内;酮康唑5-75mg/kg,腹膜内)的阿普唑仑和酮康唑预处理后的自身给药。显然,当单独施用时,这些剂量不会显著影响可卡因自身给药(或食物维持反应)。“组合”(小条纹)柱描绘了在递送组合药物疗法(即由无效剂量的阿普唑仑和酮康唑组成的注射剂)后自身给药的可卡因输注的数量。可以清楚地看出,由阿普唑仑和酮康唑组成的组合药物疗法将可卡因自身给药减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到消退水平,而不影响食物维持反应。这表明所述组合降低了寻求可卡因的动机而不影响对另一种强化物(即食物)的反应或动机。这些数据还展示,用至少两种不同的皮质酮合成抑制剂和两种不同的苯并二氮杂卓观察到组合药物疗法的作用。
图4显示了设计来研究在训练不同组大鼠来自身给药不同剂量的可卡因时无效剂量的酮康唑(例如12.5mg/kg,腹膜内)和阿普唑仑(例如1mg/kg,腹膜内)的组合对可卡因自身给药的作用的实验的结果。出于与上文所讨论的用甲吡酮和奥沙西泮进行的研究中所提供的相同的原因,这是重要的。x轴上的数字表示自身给药的可卡因的三种剂量。“载体”显示当递送载体(0.9%盐水中的5%乳化剂)时自身给药的输注数量。“keto12.5”描绘了递送无效剂量的酮康唑(即12.5mg/kg,腹膜内)后自身给药的可卡因输注的数量,而“alp1”表示递送无效剂量的阿普唑仑(即1mg/kg,腹膜内)后自身给药的可卡因输注的数量。“keto/alp”表示用无效剂量的酮康唑和阿普唑仑的组合预处理后自身给药的可卡因输注的数量。显然,无论可用于自身给药的可卡因剂量如何,这种组合都显著减少了可卡因自身给药。这指示组合药物疗法的作用不会因为增加可卡因的摄入或剂量而被容易地克服。
酮康唑和奥沙西泮对静脉内自身给药可卡因的作用:这些实验被设计来确定酮康唑和奥沙西泮的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。参考图5,实心柱(“veh”)描绘了用治疗药物的载体(0.9%盐水中的5%乳化剂)预处理后自身给药的可卡因输注的数量。空心柱(“ext”)显示当对“有效”杆反应仅引起盐水输注时的消退结果。条纹柱(“ox”)和阴影柱(“keto”)表示单独用无效低剂量(奥沙西泮10mg/kg,腹膜内;酮康唑12.5mg/kg,腹膜内)的奥沙西泮和酮康唑预处理后的自身给药。当单独施用时,这些剂量不会显著影响可卡因自身给药(或食物维持反应)。小条纹柱(“组合”)描绘了在递送组合药物疗法(即由无效剂量的奥沙西泮和酮康唑组成的注射剂)后自身给药的可卡因输注的数量。如图5中可见,由奥沙西泮和酮康唑组成的组合药物疗法将可卡因自身给药减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到消退水平而不影响食物维持反应,表明该组合减少了寻求可卡因的动机而不影响另一种强化物(即食物)的反应或动机。这些数据还展示,用不同的皮质酮合成抑制剂和不同的苯并二氮杂卓观察到组合药物疗法的作用。
cp-154,526和奥沙西泮自身给药对静脉内给药可卡因的作用:
这些实验被设计来确定cp-154,526和奥沙西泮的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。结果在图6中以测试条件下基线输注的百分比的形式呈现。白色柱(“ext”)显示当对“有效”杆反应仅引起盐水输注时的消退结果。标记为“cp-高”的柱描绘了用高剂量cp-154,526(10-80mg/kg,腹膜内)预处理后自身给药的输注的数量,而“ox-高”柱描绘了用高剂量的奥沙西泮(5-25mg/kg,腹膜内)预处理后自身给药的可卡因输注的数量。这些剂量下的cp-154,526和奥沙西泮两者都减少了可卡因自身给药,而不影响食物维持反应。“cp-低”和“ox-低”柱表示单独用无效低剂量(cp-154,526,5-25mg/kg,腹膜内;奥沙西泮,5-25mg/kg,腹膜内)的cp-154,526和奥沙西泮预处理后的反应。当单独施用时,这些剂量不会显著影响可卡因自身给药(或食物维持反应)。“com-低”柱描绘了在递送组合药物疗法(即由无效剂量的cp-154,526和奥沙西泮组成的注射剂)后自身给药的可卡因输注的数量。如图6中可见,由cp-154,526和奥沙西泮组成的组合药物疗法将可卡因自身给药减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到消退水平而不影响食物维持反应,表明该组合减少了寻求可卡因的动机而不影响另一种强化物(即食物)的反应或动机。这些数据还展示,用苯并二氮杂卓和crh受体拮抗剂的组合观察到组合药物疗法的作用。
甲吡酮和阿普唑仑对静脉内自身给药可卡因的作用:这些实验被设计来确定甲吡酮和阿普唑仑的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。参考图7,最左边的柱(“veh”)描绘了用治疗药物的载体(0.9%盐水中的5%乳化剂)预处理后自身给药的可卡因输注的数量。标记“ext”的柱显示当对“有效”杆反应仅引起盐水输注时的消退结果。“met-h”柱显示用高剂量甲吡酮(25-175mg/kg,腹膜内)预处理后自身给药的可卡因输注的数量,而“alp-h”柱描绘了用高剂量阿普唑仑(1-5mg/kg,腹膜内)预处理后自身给药的可卡因输注的数量。这些剂量下的甲吡酮和阿普唑仑都减少了可卡因自身给药,而不会影响食物维持反应。“met-l”和“alp-l”柱表示单独用无效低剂量(甲吡酮,25-50mg/kg,腹膜内;阿普唑仑0.5-2mg/kg,腹膜内)的甲吡酮和阿普唑仑预处理后的反应。当单独施用时,这些剂量不会显著影响可卡因自身给药或食物维持反应。“组合”柱描绘了在递送组合药物疗法(即由无效剂量的甲吡酮和阿普唑仑组成的注射剂)后自身给药的可卡因输注的数量。由甲吡酮和阿普唑仑组成的组合药物疗法将可卡因自身给药减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到消退水平而不影响食物维持反应,表明该组合减少了寻求可卡因的动机而不影响另一种强化物(即食物)的反应或动机。
蝇蕈醇和cp-154,526对静脉内自身给药可卡因的作用:这些实验被设计来确定cp-154,526和蝇蕈醇的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。结果如图8所示。标记“ext”的柱描绘了当对“有效”杆反应仅引起盐水输注时的消退结果。“mus-高”柱显示用高剂量蝇蕈醇(1-4mg/kg,腹膜内)预处理后自身给药的输注的数量,而“cpx-高”柱描绘了用高剂量的cp-154,526(10-80mg/kg,腹膜内)预处理后自身给药的可卡因输注的数量。这些剂量下的蝇蕈醇和cp-154,526两者都减少了可卡因自身给药,而不会影响食物维持反应。“mus-低”和“cp-低”柱表示单独用无效低剂量蝇蕈醇(0.5-2.0mg/kg,腹膜内)和cp-154,526(5-25mg/kg,腹膜内)预处理后的反应。当单独施用时,这些剂量不会显著影响可卡因自身给药或食物维持反应。“com-低”柱描绘了在递送组合药物疗法(即由无效剂量的蝇蕈醇和cp-154,526组成的注射剂)后自身给药的可卡因输注的数量。如图8中可见,由蝇蕈醇和cp-154,526组成的组合药物疗法将可卡因自身给药减少到接近在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到接近消退水平而不影响食物维持反应,表明所述组合减少了寻求可卡因的动机而不影响另一种强化物(即食物)的反应或动机。
蝇蕈醇和甲吡酮对静脉内自身给药可卡因的作用:这些实验被设计来确定蝇蕈醇和甲吡酮的组合对在食物强化和可卡因自身给药的多重交替方案下反应的大鼠中静脉内可卡因自身给药的作用。结果如图9所示。标记“ext”的柱描绘了当对“有效”杆反应仅引起盐水输注时的消退结果。“mus-高”柱显示用高剂量蝇蕈醇(1-4mg/kg,腹膜内)预处理后自身给药的输注的数量,而“met-高”柱描绘了用高剂量的甲吡酮(25-175mg/kg,腹膜内)预处理后自身给药的可卡因输注的数量。这些剂量下的蝇蕈醇和甲吡酮两者都减少了可卡因自身给药,而不会影响食物维持反应。“mus-低”和“met-低”柱表示单独用无效低剂量蝇蕈醇(0.5-2.0mg/kg,腹膜内)和甲吡酮(25-50mg/kg,腹膜内)预处理后的反应。当单独施用时,这些剂量不会显著影响可卡因自身给药或食物维持反应。“com-低”柱描绘了在递送组合药物疗法(即由无效剂量的蝇蕈醇和甲吡酮组成的注射剂)后自身给药的可卡因输注的数量。如图9中可见,由蝇蕈醇和甲吡酮组成的组合药物疗法将可卡因自身给药减少到接近在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将可卡因自身给药减少到接近消退水平而不影响食物维持反应,表明gabaa受体激动剂和皮质酮合成抑制剂的组合减少了寻求可卡因的动机而不影响另一种强化物(即食物)的反应或动机。
长期注射甲吡酮对线索诱导的已消退可卡因寻求行为的恢复的作用:这些实验被设计来确定长期施用甲吡酮对大鼠中线索诱导的已消退可卡因寻求的恢复的作用。使用可卡因寻求的复发模型。这是一项重要的实验,因为组合药物疗法将被长期施用于可卡因成瘾者。参考图11,标记为“sa”的柱描绘了在可卡因自身给药期间对“有效”杆反应的次数。标记为“ext”的柱描绘了在消退期间当对“有效”杆反应仅引起盐水输注时对这个杆的反应次数。标记“veh”的柱表示在恢复测试期间在用载体(0.9%盐水中的5%乳化剂)预处理后对“有效”杆的反应。标记为“甲吡酮”的柱描绘了在恢复测试期间在长期递送甲吡酮(50mg/kg,腹膜内,每天一次,持续14天)后对“有效”杆反应的次数。如图11中可见,长期施用甲吡酮将可卡因寻求减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。这些数据展示长期施用后甲吡酮仍有效阻断可卡因寻求的复发。
cp-154,526和奥沙西泮的组合对线索诱导的已消退可卡因寻求行为的恢复的作用:这些实验被设计来确定cp-154,526和奥沙西泮的组合对大鼠中线索诱导的已消退可卡因寻求的恢复的作用。参考图12,标记“自身给药”的该组柱描绘了在可卡因自身给药期间对“有效”杆和第二“无效”杆作出的反应的次数。该组中的前两个柱表示在恢复测试期间最终接受载体(0.9%盐水中的5%乳化剂)作为治疗药物的大鼠的反应。第三和第四柱描绘了在恢复测试期间最终接受组合药物疗法(即由无效剂量的cp-154,526和奥沙西泮组成的注射剂,如可卡因自身给药实验中所确定)的大鼠的反应。在恢复测试训练期开始前30分钟,仅向大鼠注射载体或组合药物疗法一次。自身给药和消退期间的反应仅在图12中呈现以展示组之间的反应没有显著差异。对“无效”杆的反应在任何时候都不产生程序化结果。标记“消退”的第二组柱描绘了在消退期间在对“有效”杆反应仅引起盐水输注时对“有效”和“无效”杆反应的次数。标记“恢复”的第三组柱描述了在恢复测试期间在递送组合药物疗法(即由无效剂量的cp-154,526和奥沙西泮组成的注射剂,如在可卡因自身给药实验中所确定)后对“有效”和“无效”杆反应的次数。明显可见,由cp-154,526和奥沙西泮组成的组合药物疗法将可卡因寻求(即在恢复期间对有效杆的反应)减少到在消退期间当按压有效杆时仅递送盐水时所见的水平。组合药物疗法将恢复(复发)减少到消退水平,而不影响食物维持反应。这表明该组合降低了寻求可卡因的动机而不影响对另一种强化物(即食物)的反应或动机。
没有可卡因、甲吡酮和奥沙西泮之间药代动力学相互作用的证据:向成年雄性wistar大鼠(90至120天龄)植入长期留置颈静脉导管并使其从手术中恢复。在测试当天,在施用可卡因注射剂前30分钟,用腹膜内注射奥沙西泮和甲吡酮的各种组合(如图14的表中所示)或载体(盐水中的5%乳化剂)预处理大鼠。奥沙西泮/甲吡酮组合选自行为研究,所述研究展示这些组合减少了可卡因自身给药或线索诱导的已消退可卡因寻求的恢复,而不影响食物维持反应。在药物组合或载体注射后30分钟,大鼠每2分钟接收静脉内注射可卡因(每次输注0.25mg/kg/),持续1小时。在最后一次注射可卡因后,从导管中收集血液用于分析可卡因以及其代谢物芽子碱甲酯和苯甲酰芽子碱。还测定了甲吡酮和甲吡醇以及奥沙西泮的浓度。使用gcms程序测定所有药物浓度。这些研究的结果展示奥沙西泮和甲吡酮的组合对可卡因或其代谢物的血浆浓度无作用。这些研究还展示甲吡酮和奥沙西泮不会影响彼此的血浆浓度。此外,可卡因的存在不影响甲吡酮或奥沙西泮的血浆浓度。这些数据表明在大鼠中观察到的行为作用不是因为各种药物之间的药代动力学相互作用。
在强迫游泳测试——抑郁的动物模型中测试的奥沙西泮和甲吡酮的组合:强迫游泳测试(fst)是一种具有用于评定药物抗抑郁功效的预测有效性的动物模型。将受试者暴露于不可避免的危及生命的情形来引发习得性无助。为了达到这个目的,将大鼠放入装满水的圆筒中,它们无法从中逃脱并且必须游泳来保持漂浮。在大鼠“意识到”其情形毫无希望时,出现类似绝望的行为,并且大鼠变得不动而不是尝试逃脱或游泳。这种不动姿势的时间是测量为绝望的行为。奥沙西泮(一种苯并二氮杂卓)和甲吡酮(一种皮质酮合成的11-β抑制剂)已显示分别具有抗焦虑和抗抑郁功效。使用fst在雄性wistar大鼠中评估单独和共同、短期和长期地施用的奥沙西泮和甲吡酮的潜在抗抑郁特性。在测试后第一天和再次在测试之前第二天(短期)或在第一天开始测试之前的14天(长期),向大鼠注射一种药物(5或10mg/kg奥沙西泮、25或50mg/kg甲吡酮)或其组合。短期和长期、单独和组合地施用药物有效减少fst中的不动,指示这种药物疗法具有抗抑郁活性。
习得性无助是使用fst作为抑郁模型的有效性所基于的构想。在人类中,习得性无助往往表现为抑郁的症状,这表现为丧失应对能力。出于这个原因,假设具有减少fst中不动时间的作用的药物具有潜力作为减轻人类抑郁模型中所见的应对能力丧失的候选物。在其它研究中,在fst中单独和组合地测试了奥沙西泮和甲吡酮来判定这些药物是否可以显示出抗抑郁活性。
研究的参数如上所概述。更具体地,使用来自harlan的体重为275-400克的雄性wistar大鼠。在测试之前,允许大鼠在到达之后在动物资源设备(animalresourcesfacility)中适应至少一天。为了进行fst,用新鲜的25℃水填充plexiglas圆筒(高度40cm×直径18cm)到20cm的深度,该深度足够深,使得大鼠不能触及底部,但距离边缘足够远而防止大鼠逃脱。在测试后第一天和在测试之前第二天(短期)再次或在第一天开始测试之前的14天(长期),向大鼠腹膜内注射载体、药物或奥沙西泮和甲吡酮的组合。在第一天,将大鼠从笼中取出,置于水中,并观察15分钟。一般来说,对于最初的几分钟,大鼠会四处游动,并用其爪在水位线上方划动,嗅闻,潜水,并尝试从圆筒中跳出。这动作被视为导向逃脱行为。在导向逃脱行为之后是特征为大鼠停止其逃脱尝试的时间。通常,老鼠会踩水,只施加足以使其头部保持在水面之上的能量,或会漂浮而只使其鼻子高于水位线。这第二阶段的行为被认为是不动姿势。记录在导向逃脱行为和不动姿势中花费的时间长度。然后将大鼠从水中取出,用毛巾擦干,并返回其居住的笼中。在第二天,重复该程序五分钟,并记录从事导向逃脱行为和不动姿势所花费的时间。比较了不同组之间第二天的不动持续时间。使用单因子anova将剂量组与注射载体的对照进行比较,其中p<0.05。如果药物组的不动时间与载体组相比具有统计学显著性,则认为药物组合展现抗抑郁样作用。
在组合治疗组中,长期施用奥沙西泮和甲吡酮的作用更显著。只有一个施用单独药物的组——met50组显示不动时间减少。这表明当两种药物同时施用时具有协同作用。这种协同作用也许可以通过奥沙西泮对由甲吡酮的代谢副产物所诱导的gabaa受体的激动作用增加来解释。当甲吡酮抑制皮质酮合成时,皮质酮上游的两种前体——11-脱氧皮质酮(11-doc)和孕酮(prog)的浓度增加。这种增加可以将路径分流至产生gabaa活性神经类固醇如别孕烯醇酮和四氢脱氧皮质酮。这两种神经类固醇以别构方式与gabaa受体结合,使得cl-流入细胞增加,从而引起超极化并降低神经元兴奋性。可能的结果是奥沙西泮(通过直接结合)和甲吡酮(间接地通过神经类固醇)两者都通过别构机制影响gabaa电流。无论作用机制如何,很明显ox10/met50的组合引发了不动时间的最大减少。在长期处理组中似乎已经形成了耐受,特别是仅接受ox或met的那些组。这一点通过这些组的平均值等于或超过载体的观察结果而显而易见。
实施例2
这个实验被设计来评定甲吡酮(met)和奥沙西泮(ox)的组合在人类中的安全性和药代动力学。这个实验中施用的met/ox组合(在本文中称为emb-001)是甲吡酮(met)(一种皮质醇合成抑制剂)和奥沙西泮(ox)(一种苯并二氮杂卓)的组合。met经fda批准仅使用一天来用于垂体功能的测试,并且ox经批准用于短期和长期治疗各种焦虑症。两种药物目前都未被批准用于治疗成瘾或物质滥用障碍。在先前的动物研究中,emb-001在大鼠中减少了可卡因和烟碱的自身给药,并减弱了可卡因和甲基安非他命线索反应性。在可卡因依赖性受试者的人类研究中,emb-001显著减少了可卡因的使用。
方法:这是一项单次和多次上升剂量的研究。吸烟的18-65岁健康志愿者在第1天接受单次am剂量,在第3-9天接受bid给药,并且在第10天接受单次am剂量。三个具有8名受试者(6种药物,2种安慰剂)的连续剂量组分别接受以下剂量的met和ox:270和12mg;540和24mg;以及720和24mg。在bid给药日,每日总剂量是这些量的两倍。主要结果是met、其活性代谢物(甲吡醇)和ox的安全性和药代动力学。安全性测量包括生命体征、ecg和标准的安全性实验室指标。在整个研究中密切监测皮质醇和其它hpa轴参数。
结果:最常见的不良事件是嗜睡。大多数不良事件是轻度的,并且所有都是轻度或中度的。没有严重的不良事件,也没有因不良事件而中断。与met的已知药理学一致,第一次给药后2-4小时血清皮质醇减少,但在次日早晨和随访时恢复到基线水平。生命体征、ecg或其它安全性实验室指标在临床上没有显著变化。剂量组2中的一名受试者相对于筛选经历了早晨皮质醇减少>50%,但是无症状。将研究药物停用一天(第8天),在此期间acth刺激测试揭示足够的肾上腺反应。恢复给药并且受试者完成研究。met、ox和甲吡醇的半衰期分别约为2、7.5和8小时。暴露随着剂量增加而增加。反复给药存在适度的累积。
结论:emb-001在本研究中具有良好的耐受性,并且未鉴定新的安全性信号。药代动力学结果表明,每日两次给药可以提供适当的暴露持续时间以有效治疗物质滥用障碍和其它成瘾。
实施例3
表1
实施例4
表2
实施例5
表3
安全性结果:hpa实验室结果、体征和症状
·评估整个研究中的皮质醇和acth。
·虽然一些受试者经历皮质醇减少,但没有一个展现出需要中断研究药物或治疗的肾上腺功能不全的症状。
·剂量组2中的一名受试者相对于筛选经历了早晨皮质醇减少>50%。该受试者无症状。将研究药物停用一天(第8天),随后的acth刺激测试揭示足够的肾上腺反应。
·第二天恢复给药并且受试者完成研究。
·每日肾上腺功能不全检查清单(airc)反应未显示临床上显著的病征或症状。
·在给药后2-4小时,皮质醇以剂量依赖性方式减少,但在第二天早晨和bid给药一周后的早晨恢复正常。
实施例6
表4
已经描述了本发明的多个实施方式。然而,应理解,在不脱离本发明的精神和范畴的情况下,可以进行各种修改。因此,其它实施方式在权利要求书的范畴内。
- 还没有人留言评论。精彩留言会获得点赞!