含有高分子化药物的医药组合物的制作方法
本发明基于日本特许申请第2016-93407号主张优先权,由于在此进行参照,将其全部引入本说明书中。
本发明涉及一种含有高分子化药物的医药组合物,特别是注射剂及其制造方法。
背景技术
现有的抗癌剂(也称为抗肿瘤剂、抗癌剂)的分子量大多为1500以下,并且在大多数情况下,它们在体内均匀地扩散分布,因此伴随有对正常器官的副作用,并且对于肿瘤的选择毒性很差。因此,使用这些抗癌剂,由于副作用增加,难以为了期望更强的效果而增加给药量。
对应于这种现有的一般理论,本发明人发现下述现象:通过将抗癌剂与具有生物相容性的聚合物键合等手段进行高分子化,抑制药物从血液中经由肾脏而排泄(消失),同时延长在血液中的滞留性,并利用固体肿瘤具有的增强的血管通透性(渗透性)的状态,使该高分子化药物更有选择性地在肿瘤局部向血管外渗出。本发明人等已经将该现象作为epr(高通透性和滞留,enhancedpermeabilityandretention)效应这样的一般概念(非专利文献1~4)进行了报告。
本发明人等已经开发出了,新型吡柔比星(thp)的高分子键合型抗癌剂,例如,聚羟丙基甲基丙烯酰胺(hpma)键合thp(p-thp)、苯乙烯-马来酸共聚物(sma)键合thp(sma-thp)、以及同样的zn-原卟啉(znpp)的高分子键合体(p-znpp和sma-znpp)(专利文献1~2)。另一方面,本发明人等进一步揭示了参与增强epr效应的各种药理学因素。这样的因素例如有,缓激肽、一氧化氮(no)、一氧化碳和促进这些生成的物质,ace(血管紧张素转换酶)抑制剂等(非专利文献5~8)。与肿瘤局部的no浓度相关的no释放剂及ace抑制剂,将肿瘤的epr效应提高2~3倍,同样地将上述高分子化药物向肿瘤的输送(delivery)提高2~3倍(非专利文献5~8)。
现有技术文献
专利文献
专利文献1:wo2013/035750
专利文献2:wo2015/076312
非专利文献
非专利文献1:y.matsumura&h.maeda,cancerres.(1986)46,p.6387-6392
非专利文献2:h.maeda,adv.inenzymeregulation(2001)41,p.189-207
非专利文献3:h.maedaetal,adv.drugdeliv.res(2013)65,p.71-79
非专利文献4:proc.japanjapanacad.ser.b(2012)88,pp.53-71
非专利文献5:t.sekietal,cancersci.(2009)100,p.2426-2430
非专利文献6:j.fangetal,addreview(2011)63,p.136-151
非专利文献7:h.maeda,j.controlrelease(2012)164,p.138-144
非专利文献8:fangetal,j.control.release(2016)223,p.188-196
技术实现要素:
发明要解决的技术问题
尽管大多高分子化药物,特别是由于具有复杂的高级功能结构或分子间相互作用而表现出相对均匀的分子量分布的高分子化药物,高分子分子间相互作用很强,作为结果,导致进一步形成复合体等,或由于复杂的侧链分子间的相互作用受到抑制等而不能维持高级结构,高分子化药物分子相互缔合形成凝聚块,因此存在稳定性低这样的问题。有时存在分子间的缔合过强,而不会分散在水溶液中而无法溶解。
通过分子间的缔合而具有高分子形态的胶束和脂质体制剂中,稳定性是一个特别严重的问题。例如,当胶束或脂质体制剂的溶液中的稳定性存在问题时,可能会丧失胶束形成能力或可能释放(游离)内含的药物。
因此,期望通过克服高分子化药物的溶解度及在溶液中的不稳定性,来改善这样的巨大分子在生物体内的稳定性,从而维持由epr效应产生的肿瘤选择性。此外,如果可以在这些高分子化药物进行静脉内给药时提高epr效应,则可以进一步增强抗癌效应,并减少副作用,故较为重要。
此外,在大多高分子化药物中,例如胶束制剂或所谓的纳米药物,当粉末(固体)状态溶解在水性溶剂中并用作注射剂等时,其溶解度通常较差,而在临床实践中造成问题。
解决技术问题的技术手段
本发明人等鉴于上述问题进行了深入研究,结果发现,当将高分子化药物(例如,p-thp)溶解在水性溶剂中并制备注射剂时,通过混合指定的溶解促进剂和/或稳定剂,可以缩短高分子化药物在水性溶剂中的溶解时间,即促进高分子化药物的溶解,可以使水溶液中的该药剂分子中的酯键、腙键或某些酰胺键稳定,此外,可以通过指定的溶解促进剂和/或稳定剂(例如,作为生物体内no合成酶的原料的精氨酸),实现上述高分子化药物的溶解促进和稳定化,还可以实现epr效应的增强等,从而完成了本发明。
因此,本发明包含以下内容。
[1]一种医药组合物,其包含高分子化药物及水性溶剂,并包含溶解促进剂和/或稳定剂,其中,
该溶解促进剂和/或稳定剂为选自下述中的至少一种:
(1)蛋白质、
(2)合成聚合物、
(3)糖或糖醇、
(4)无机盐类、
(5)氨基酸、
(6)磷脂、
(7)脂肪醇、
(8)中链脂肪酸、以及
(9)粘多糖。
[2]根据上述[1]所述的医药组合物,其ph为7.0~8.0。
[3]根据上述[1]或[2]所述的医药组合物,其为注射剂。
[4]根据上述[1]~[3]中任一项所述的医药组合物,其中,高分子化药物为选自p-thp、p-znpp、sma-thp、sma-znpp、peg-thp和peg-znpp中的至少一种。
[5]根据上述[1]~[4]中任一项所述的医药组合物,其中,高分子化药物中的药物和高分子之间的键为选自酰胺键、酯键、腙键和希夫氏碱键中的至少一种。
[6]根据上述[5]所述的医药组合物,其中,高分子化药物中的药物和高分子之间的键为腙键。
[7]根据上述[1]~[6]中任一项所述的医药组合物,其中,溶解促进剂和/或稳定剂为选自精氨酸和瓜氨酸中的至少一种。
[8]根据上述[1]~[7]中任一项所述的医药组合物,其还包含epr效应和/或抗肿瘤效应增强剂。
[9]根据上述[1]~[8]中任一项所述的医药组合物,其用于抗癌或抗肿瘤。
[10]根据上述[1]~[9]中任一项所述的医药组合物的制造方法,其包括:对高分子化药物以及、溶解促进剂和/或稳定剂以及水性溶剂进行混合的工序。
[11]一种医药组合物,其包含高分子化药物,并包含溶解促进剂和/或稳定剂,其中,该溶解促进剂和/或稳定剂为选自下述中的至少一种:
(1)蛋白质、
(2)合成聚合物、
(3)糖或糖醇、
(4)无机盐类、
(5)氨基酸、
(6)磷脂、
(7)脂肪醇、
(8)中链脂肪酸、以及
(9)粘多糖。
[12]根据上述[11]所述的医药组合物,其还包含epr效应和/或抗肿瘤效应增强剂。
[13]一种医药组合物,其包含高分子化药物,并包含epr效应和/或抗肿瘤效应增强剂,其中,该epr效应和/或抗肿瘤效应增强剂为选自下述中的至少一种:
(1)硝酸甘油、
(2)精氨酸、
(3)羟基脲、
(4)亚硝基脲。
[14]根据上述[13]所述的医药组合物,其还包含溶解促进剂和/或稳定剂。
[15]一种高分子化药物的溶解促进和/或稳定化的方法,其包括:将溶解促进剂和/或稳定剂混合在高分子化药物中,该溶解促进剂和/或稳定剂为选自下述中的至少一种:
(1)蛋白质、
(2)合成聚合物、
(3)糖或糖醇、
(4)无机盐类、
(5)氨基酸、
(6)磷脂、
(7)脂肪醇、
(8)中链脂肪酸、以及
(9)粘多糖。
发明的效果
根据本发明的医药组合物,可以通过指定的溶解促进剂和/或稳定剂,使高分子化药物(例如,p-thp)在水性溶剂中的溶解时间显著缩短,即可以促进高分子化药物的溶解,此外,可以使高分子化药物在溶液中的稳定性得到显著改善。此外,根据本发明,除上述效果之外,通过指定的溶解促进剂和/或稳定剂可以显着增强高分子化药物的epr效应、向肿瘤的输送(tumourdelivery)、抗肿瘤效应等。
因此,例如当使用抗肿瘤剂作为药物时,本发明的医药组合物作为抗肿瘤用医药组合物是显著优异的。此外,例如当使用荧光分子作为药物时,与给药单一药剂作为对于肿瘤的荧光探针相比,通过以本组合物的形式给药,可以促进epr效应的增强,从而获得更高的肿瘤积聚,故为非常有用的。
因此,根据本发明,作为高分子化药物的剂型,可以制成:在使用时加入指定的水溶液等时,可以容易地进行溶解的注射剂,并且,不仅能够提高注射剂的稳定性,还可以增强高分子化药物的epr效应、向肿瘤的输送、抗肿瘤效应等,因此可以增强其治疗效果并减少副作用。
此外,根据本发明,通过将指定的epr效应和/或抗肿瘤效应增强剂与高分子化药物组合给药,可以显著增强高分子化药物的epr效应、向肿瘤的输送(tumourdelivery)、抗肿瘤效应等。
附图说明
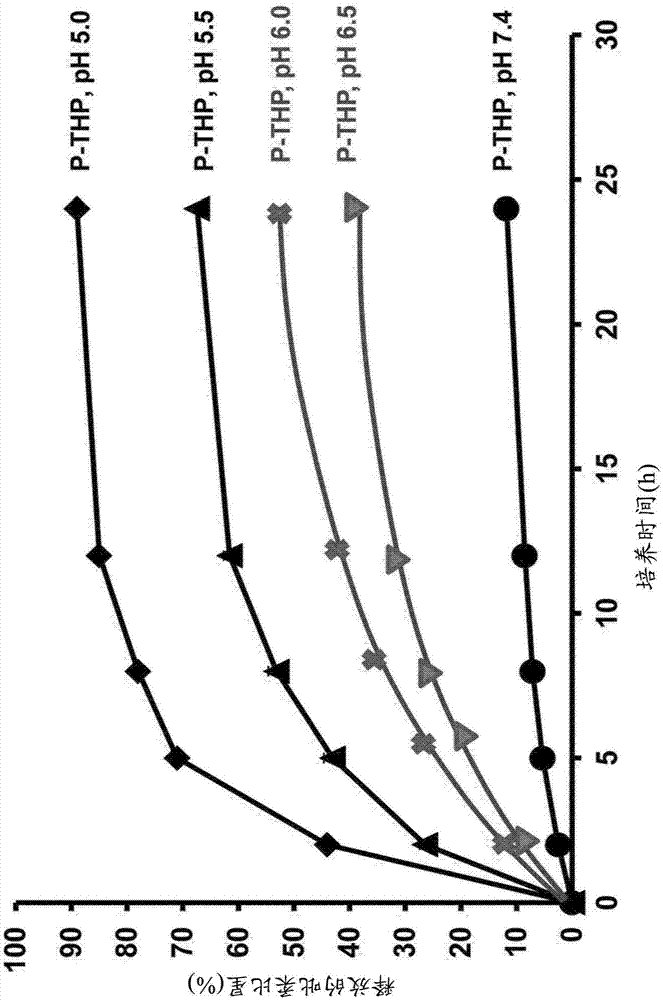
图1表示聚羟丙基甲基丙烯酰胺(hpma)键合thp(p-thp)的ph依赖性的吡柔比星的释放。
图2表示各药物对p-thp冻干物的水溶性的影响。
图3表示通过hplctsk3000柱,对聚合物键合抗癌剂p-thp(腙键)在各溶液、ph、温度条件下(a-g)的稳定性进行分析的结果。纵轴表示基于分解产生的吡柔比星(thp)的量(吸收488nm)。图3a表示3%精氨酸缓冲液的结果,与1%精氨酸缓冲液的数据大致相同。另外,图3e~图3g表示0.3m精氨酸-精氨酸hcl缓冲液(ph8.5)的结果,与3%碳酸氢钠-碳酸钠缓冲液(ph8.5)的数据大致相同。
图4表示通过sephacryls-300对p-thp进行分析的结果。洗脱溶液:a是0.1m碳酸氢钠(ph8.2)、b是3%精氨酸(ph8.5)、c是pbs(0.01m磷酸,0.15nacl,ph7.4)缓冲液。a、b均为尖锐的单峰,表示单一的分子量分布,但在pbs中,如图4c所示,进行thp的释放,附于峰上的箭头的部位表示峰的均匀性的偏离(分解物质的生成)。可发现,峰的宽度比a、b的宽度宽。
图5表示通过硝酸甘油增强p-thp对肿瘤(晚期乳腺癌)的抗肿瘤效应。
图6表示通过硝酸甘油增强p-thp对s180移植小鼠的抗肿瘤效应。
图7表示通过各epr效应和/或抗肿瘤效应增强剂增强p-thp对氧化偶氮甲烷诱导的小鼠结肠癌的抗肿瘤效应。
具体实施方式
本发明中的“高分子化药物”没有特别限定,其为由抗癌剂等药物和生物相容性聚合物形成的物质,可举出通过共价键或非共价键的键合体或复合体。
本发明中的“药物”没有特别限定,例如,可举出:新制癌菌素(ncs)、吡柔比星(thp)、zn-原卟啉(znpp)等抗癌剂、玫瑰红、亚甲蓝、吖啶、吖啶黄、吖啶橙及吲哚花青绿等荧光分子,优选举出thp及znpp等抗癌剂。
作为上述生物相容性聚合物,例如,可列举:聚羟丙基甲基丙烯酰胺(hpma)聚合物、苯乙烯-马来酸共聚物(sma)及聚乙二醇(peg)等,优选hpma聚合物及sma。
通常,作为以共价键连接上述药物与上述生物相容性聚合物的化学键,可举出:通过酰胺(r1-co-nh-r2:其中,r1和r2是任意基团,以下相同)、酯(r1-co-o-r2)、醚(r1-o-r2)、二硫化物(r1-s-s-r2)、腙(r1-co-nnh-r2)、希夫氏碱(-c=nh-)的键、腙(肼)键等。
特别是,酰胺和酯被广泛使用,腙键由于具有弱酸环境响应性(药物的释放性)的功能而被广泛使用(f.kratzetal,drugdeliv.6,89-95(1999);bioorganicmed.chem.lett.7,617-622(1997)等)。
此外,在酰胺键中,当r1是马来酸、乌头酸与药物(例如抗癌剂化合物)的氨基之间的键合时,药物在弱酸性ph下或0.1%sds(十二烷基硫酸钠)的存在下释放。
作为上述药物与生物相容性聚合物的键合体/复合体,例如wo/2003/018007、wo2004/103409、wo2006/112361、wo2013/035750、wo2015/076312等中记载的键合体/复合体、例如可举出p-thp、p-znpp、sma-znpp、sma-thp、peg-thp、peg-znpp、sma-cddp等。优选举出下述表1中所记载的。
[表1]
表1、本发明的静脉注射剂中使用的高分子键合药剂的示例
具体而言,可举出由以下化学式表示的键合体。
(1)sma键合thp(sma-thp复合体,酰胺键)
[化学式1]
(2)hpma-聚合物-thp(吡柔比星)(称为“p-thp”)(腙键)
[化学式2]
(3)sma-共聚物-thp(腙键)
[化学式3]
(4)hpma-znpp
[化学式4]
(5)poly-hpma-znpp
[化学式5]
(6)sma-znpp
[化学式6]
上述键合体(1)和(2)优选满足下述性质中的任一项,特别优选满足下述全部性质。
·分子量(mw):>40kda
·尺寸/dls:~50nm
·表面电荷:-28mv
·thp负载量:1~50%,优选10%(w/w)
·细胞摄取:dox的>×10~×100
·等离子体t1/2:thp的100~200倍
·dl50:100~200mg/kg(与原药剂相比~10倍良好:即低毒性)
·肿瘤/血液:thp的>110~200
·体外细胞毒性:游离thp的0.5~50%
上述键合体在生物体内的稳定性根据化学键的种类的不同有很大不同,在血清成分的存在下,容易以醚、酰胺、酯、肼键的顺序发生分解。另一方面,腙在ph较低的情况下最易于切断。
此外,在动物血清中对于酯键的切断很大程度上取决于动物的差异,按照小鼠、大鼠>兔>人的顺序变慢。此外,在人大肠癌的匀浆中,比在正常组织中更快地被切断,并且按照酯>酰胺>醚的顺序变慢(tsukigawaetal,eur.j.pharm.biopharm89,259-270(2015))。
就上述键合体(1)~键合体(3)而言,可以由thp分子内存在的氨基、羧基或酮基、及sma的马来酸酐基或羧基、或hpma的羟基,或者通过wo2015/076312等中提及的腙等连接,形成酯键、酰胺键或腙键,制成键合体。
这样的键合体,例如,如wo2013/035750、wo2015/076312、h.nakamuraetal,j.controlrelease(2014)174,p81-87、h.nakamuraetal,j.controlrelease(2013165,p191-198等中记载的,并且可以通过这些文献中记载的方法来制造。
作为用于本发明的“溶解促进剂和/或稳定剂”没有特别限定,只要可以改善上述高分子化药物在水性溶剂中的溶解性和/或稳定性即可,例如可举出下述物质:
(1)蛋白质:人血清白蛋白、转铁蛋白、免疫球蛋白、可溶性明胶、琥珀酰化(酰化)明胶、改性明胶等;
(2)合成聚合物:聚乙二醇(peg)、聚丙二醇、乙烯醇、聚乙烯醇、聚乙烯吡咯烷酮、羟丙基甲基丙烯酰胺(hpma)聚合物等;
(3)糖或糖醇:甲基溶纤剂、甘草酸、葡萄糖、甘露醇(mannitol)、麦芽糖、山梨糖醇、山梨酸、乳糖、海藻糖、葡聚糖、环糊精、甘油(glycerol)、可溶性淀粉;
(4)无机盐类:碳酸氢钠等;
(5)氨基酸:甘氨酸、甘氨酰甘氨酸、丙氨酸、丝氨酸、苏氨酸、谷胱甘肽、半胱氨酸、精氨酸(l-精氨酸)、赖氨酸、组氨酸、鸟氨酸及瓜氨酸;
(6)磷脂:卵磷脂等;
(7)脂肪醇:十六烷基醇等;
(8)中链脂肪酸:碳原子数5~10的脂肪酸,例如辛酸等;
(9)粘多糖:透明质酸、硫酸软骨素等。
这些可以是被日本药典批准的,也可以是未经批准的。此外,可以组合使用这些中的两种以上。
在本发明的医药组合物中,从提高高分子键合药物的溶解性、提高稳定性、抑制分解性、增强epr效应、向肿瘤的输送、抗肿瘤效应等观点出发,特别可举出氨基酸(精氨酸、甘氨酸、瓜氨酸等)、碳酸氢钠及peg等。
相对于上述高分子化药物1重量份,上述溶解促进剂和/或稳定剂的配合量通常为0.01~50重量份,优选为1~10重量份。
作为本发明中使用的“水性溶剂”,只要可用于注射剂等即可,没有特别限定,可举出:蒸馏水、去离子水、纯化水、灭菌净化水、注射用水等水、以及向这些水中加入添加剂而得到的物质,例如生理盐水(5%)、5%碳酸氢钠水溶液、林格氏液等。此外,其ph通常为9.0以下,优选为例如,7.8~8.7、7.0~8.0等。渗透压没有特别限定。
就通过上述键合体(1)及键合体(2)等的腙键、马来酰胺键等键合而得到的药物而言,溶液为酸性ph时,可以从聚合物中释放、放出(图1)。因此,当本发明的医药组合物是液体制剂时,ph优选为6.0以上,更优选为例如7.5~9.0、7.8~8.7、7.0~8.0等。
本发明的医药组合物可以通过制剂领域的标准方法来制造。例如,本作为注射剂等液体制剂的发明的医药组合物可通过下述方法制造:将上述高分子化药物及上述溶解促进剂和/或稳定剂溶解于10ml~1l的水性溶剂(水溶液)中,使得上述溶解促进剂和/或稳定剂相对于上述高分子化药物1g为0.01g~50g(优选为0.1g~10g)。
此外,上述高分子化药物及上述溶解促进剂和/或稳定剂的浓度可根据所期望的效果、给药方法(静脉注射、滴注剂等)适当设定。例如,作为高分子化药物的浓度,可举出0.01~60%(w/v),特别可举出0.1~20%(w/v)。此外,作为上述溶解促进剂和/或稳定剂的浓度,例如,可举出0.1~10%(w/v),特别可举出1~10%(w/v)。
本发明的医药组合物可以不含水性溶剂。即,本发明的医药组合物中,包含高分子化药物、及水性溶剂,并包含溶解促进剂和/或稳定剂的医药组合物,除此之外,还包含含有高分子化药物、溶解促进剂和/或稳定剂的医药组合物。
本发明中含有高分子化药物、溶解促进剂和/或稳定剂的医药组合物可以通过制剂领域的标准方法制造。例如,可以通过仅对高分子化药物、溶解促进剂和/或稳定剂进行混合而制造。
此外,如本发明的医药组合物还可以通过将上述液体制剂以常规方法冻干来制造。在这种情况下,本发明的医药组合物可以以固体制剂(固体注射剂)的形式,稳定地储存更长的时间。
此外,上述高分子化药物、及上述溶解促进剂和/或稳定剂可以是分别单独的固体制剂,也可以是混合物的固体制剂。能够在水性溶剂中使用的各种添加剂可以混合在这些固体制剂中。此外,还可以制成包含多种制剂的试剂盒。
上述固体制剂可以在使用时溶解在任意容量的蒸馏水中,可以溶解在少量(约10ml)蒸馏水中制成注射剂,也可以溶解在更多的蒸馏水(10ml~500ml,优选200~300ml)中制成静脉输注液。
本发明的医药组合物优选为注射剂。本发明中的“注射剂”包括水性注射剂、悬浊性注射剂、乳浊性剂注射剂、固体注射剂、静脉注射液、输液制剂等。在本发明中,优选为用于静脉注射或静脉内输注的注射剂。
另外,在本发明的医药组合物中,上述溶解促进剂和/或稳定剂可以与上述高分子化药物制成单一的制剂,也可以制成各自的制剂,同时或者分别以相同或不同的途径,向患者(人的哺乳动物)进行给药。例如,对含有上述高分子化药物的直径进行静脉给药,对上述溶解促进剂和/或稳定剂进行腹腔内给药。
就在本发明的医药组合物而言,除上述溶解促进剂和/或稳定剂之外,或者可以含有与上述溶解促进剂和/或稳定剂不同的epr效应和/或抗肿瘤效应增强剂。作为epr效应和/或抗肿瘤效效应果增强剂,只要是能够增强epr效应和/或抗肿瘤效应的药剂即可,没有特别限定,例如,可举出:(a)硝酸甘油(ng)、(b)isdn(isosorbitedinitrate)、(c)佩尔地平(perdipine)等含有硝基的降压剂、(d)苏丹类药物、(e)血管紧张素转化酶抑制剂(ace抑制剂),例如异搏定(r)(verapamil(r))(依那普利)、(f)作为血管通透性增强因子,本发明人等开发的一氧化碳co释放剂碳酸钌(corm2)的苯乙烯马来酸共聚物胶束制剂(参照j.fangetal,j.control.release(2014)187,p.14-21)、(g)具有作为co合成酶之一的heme-oxygenaze-1(ho-1)的诱导能力的氯化高铁血红素或氯化高铁血红素衍生物(例如peg化氯化高铁血红素)、(h)作为前列腺素i2的衍生物的稳定型制剂的贝前列素钠、(i)no(一氧化氮)的合成酶(一氧化氮合酶,nos)的底物,例如精氨酸(l-精氨酸)、瓜氨酸、(j)no释放剂,例如硝基吖嗪、亚硝酸、硝基戊醇、s-亚硝基-谷胱甘肽、s-硝基谷胱甘肽、s-亚硝基-半胱氨酸、(k)脲衍生物,例如羟基脲、亚硝基脲等。
优选举出isdn、硝酸甘油、佩尔地平、ace抑制剂、硝普钠、亚硝胺醇、罗沙坦类降压剂、精氨酸、羟基脲、亚硝基脲等,特别优选举出硝酸甘油、精氨酸、羟基脲等。
关于上述药剂(i),作为no合成酶(nos)的底物的精氨酸,可以在肿瘤部位生成no,通过血管扩张作用增强epr效应。通过组合使用精氨酸,可以持续维持肿瘤局部的no生成,并且可以与上述ng同样地持续增强epr效应。与精氨酸一样,瓜氨酸也可以使用,因为其在精氨酸合成循环中变为精氨酸琥珀酸酯(arginosuccinate),然后变为精氨酸,同样成为no产生的原料。
此外,对于上述药剂(j),亚硝酸根离子在氧分压较低的肿瘤部位被亚硝酸还原酶转化为no,从而提高epr效应(t.sekietal,cancerscience(2009)100,2426-2430)。
本发明的医药组合物中的epr效应和/或抗肿瘤效应增强剂的混合量没有特别限定,只要可以获得期望的效果即可,例如为1μg~100mg/安瓿。此外,当本发明的医药组合物是液体制剂时,作为其浓度,例如,可举出0.1~30(w/v),特别可举出1~10%(w/v)。
上述epr效应和/或抗肿瘤效应增强剂可以在本发明的医药组合物的制造方法的任意阶段混合。例如,可以预先溶解在上述水性溶剂中,也可以与上述高分子化药物和/或上述溶解促进剂和/或稳定剂混合,还可以与上述高分子化药物和/或上述溶解促进剂和/或稳定剂同时加入水性溶剂中。此外,更优选在对患者进行点滴时加入输注溶液(药剂)中。
此外,在本发明的医药组合物中,上述epr效应和/或抗肿瘤效应增强剂与上述高分子化药物可以制成一个制剂,或分别作为单独的制剂,同时或分别,以相同或不同的途径,向患者(人等哺乳动物)给药。例如,含有上述高分子化药物的制剂可以口服给药,上述epr效应和/或抗肿瘤效应增强剂可以通过涂布给药。
在本发明的医药组合物中,精氨酸可用作溶解促进剂和/或稳定剂,也可用作epr效应和/或抗肿瘤效应增强剂。当本发明的医药组合物是含有精氨酸的液体制剂时,精氨酸的浓度通常为0.01~30%(w/v),优选为0.1~10%(w/v),液体制剂的ph通常为7.0~9.0,优选为例如7.8~9.5、8.0~9.0、8.2~8.8附近、7.0~8.0等。此外,在该液体制剂中,可以添加葡萄糖或甘露醇0.1~10%(w/v),优选添加约8%(w/v),此外,可以添加适量的isdn、硝酸甘油、或佩尔地平(1μg~100mg/安瓿)。
本发明的医药组合物可任选地含有各种医药制剂用的添加剂,例如:ph调节剂、分散剂、润湿剂、稳定剂、防腐剂、悬浊剂、表面活性剂等。这些的用量可以通过常规方法来确定。
实施例
在下文中,将参考实施例、试验例等更详细对本发明进行说明,但本发明不限于以下实施例。
(试验例1)通过组合使用epr效应和/或抗肿瘤效应增强剂来增强p-thp的向肿瘤的输送
样品:由聚羟丙基甲基丙烯酰胺键合的吡柔比星(腙键)(p-thp)(表观分子量为40000以上)的水溶液制备而成的冻干物用作样品。
方法:将10mg各样品和特定量的表2中所示的评价药剂溶于1ml生理盐水(0.01m磷酸,0.15mnacl,ph7.4)(10mg/ml),将伊文思蓝10mg/ml水溶液以各0.1ml向至s-180小鼠(肿瘤模型)进行静脉内给药。此时,使用小鼠的肿瘤尺寸为直径5~7mm。第二天,分别解剖,取出固体肿瘤,通过常规方法提取伊文思蓝,并通过560nm的吸收来对漏出的伊文思蓝进行定量(参照非专利文献1)。其结果如表2所示。
[表2]
表2、通过组合使用epr效应增强剂对于向肿瘤输送的改善
(数值为快速静脉注射(bolus)给药后的伊文思蓝、白蛋白的相对积蓄量:药剂p-thp*)
*聚羟丙基甲基丙烯酸酰胺键合吡柔比星(腙键)
如上述表2所示,阿齐沙坦、精氨酸和硝酸甘油均显示出30%以上的显着增强效果。
(试验例2)各药剂对p-thp冻干物的溶解性的影响
样品:根据nakamura等的报道(j.controlledrelease,174,81-87(2014))来制备p-thp(腙键),然后将500mg该制备产物溶解在蒸馏水中,并通过常规方法冻干。将该冻干粉末用作样品。
方法:将10mg上述样品粉末放入各试管中,加入10ml含有下述表3和图2中所示的指定量的用于促进可溶化的试验化合物(精氨酸以下到甘氨酸为止)的溶液。这些水溶液是1~10%,0.3m(mol/l)精氨酸/精氨酸hcl缓冲液或1~5%碳酸氢钠/碳酸钠缓冲液,将它们均调节至ph8.5。然后,在振动下通过目视判定可溶度。用秒表测定目视观察的直到完全溶解的时间。其结果如下述表3和图2所示。
[表3]
表3、各药剂对p-thp冻干物的溶解性的影响(ph8.5)
(试验例3)p-thp粉末干燥制剂在各水溶液中的溶解时间
样品:根据nakamura等的报道(j.controlledrelease,174,81-87(2014))来制备p-thp(腙键),然后蒸发溶剂,得到粉末干燥物(不是冻干物)。将该粉末干燥物用作样品。
方法:将10mg上述样品(p-thp的干燥物)放入试管中,加入含有指定量的下述表4所示的精氨酸、碳酸氢钠、甘露醇、peg、或甘氨酸的水溶液(10ml,ph8.0~9.0)。需要说明的是,作为水溶液,对照以下与表3相同,使用0.3m精氨酸/精氨酸hcl缓冲液或3%碳酸氢钠/碳酸钠缓冲液(ph8.0~9.0)。
接下来,根据试验例2中所述的方法,用秒表对振动下p-thp的完全溶解时间进行测定。将其结果、试验中使用的各种水溶液、其浓度与其ph一起示于表4中。
[表4]
表4、p-thp粉末干燥物在各水溶液(10mg/ml)中的溶解时间
如表4所示,通过使用指定的精氨酸等溶解促进剂和/或稳定剂,p-thp的水溶性得到了大幅改善。
(实验例4)
样品:使用与试验例2同样地制备而得到的p-thp的冻干物作为样品。
方法:将上述样品(p-thp的冻干物)溶解在下述表5所示的各溶液中,并在下述表5所示的条件下培养。需要说明的是,作为水溶液,使用0.1m乙酸/乙酸钠缓冲液(ph6.0)、0.1m磷酸缓冲液(ph7.0、ph8.2、ph8.6)、0.3m精氨酸/精氨酸hcl缓冲液或3%碳酸氢钠/碳酸钠缓冲液(ph8.5)。
然后,通过hplc(高效液相色谱法)(柱:jskgelsw3000,检测:吸收488nm,洗脱:80%甲醇、20%0.1m乙酸钠ph7.0混合液)进行分离,通过在488nm处的吸收对作为游离的分解产物的吡柔比星(thp)进行定量,计算出原p-thp的减少量,并绘制p-thp在各溶液状态下的稳定性。其结果如表5和图3a~图3g中所示。
[表5]
图3a表示3%的精氨酸缓冲液的结果,表示出了与1%的精氨酸缓冲液基本相同的结果。此外,图3e~图3g表示0.3m精氨酸/精氨酸hcl缓冲液(ph8.5)的结果,表示出了与3%的碳酸氢钠/碳酸钠缓冲液(ph8.5)基本相同的结果。从该结果发现,在精氨酸缓冲液和碳酸氢钠缓冲溶液在ph8.5附近,p-thp表现出最佳的稳定性。
(试验例5)
样品:与试验例2同样地制备的p-thp用作样品。
方法:a溶液(实施例20:0.1m碳酸氢钠水溶液,ph8.2)、b溶液(实施例21:3%精氨酸缓冲液,ph8.5)、c溶液(比较例10:pbs(0.01m磷酸、0.15mnacl),ph7.4)分别溶于p-thp中,并在室温下静置24小时。然后,进行sephacryls300的柱色谱(φ1.8×70cm)。使用相同的溶液分别洗脱柱。
然后,利用hplc(高效液相色谱法)jskgelsw3000,并使用80%甲醇、20%0.1m乙酸钠ph7.0混合液洗脱,通过在488nm处的吸收,对上述条件下的游离的吡柔比星(thp)进行测定。其结果如图4所示。
如图4a~图4c所示,a和b溶液均显示出尖锐的单一的标准的峰,但在c溶液中,如图4c图表上的箭头所示,生成thp的分解产物,且未确认到峰的均匀性。此外,c峰的宽度比a、b的宽度宽。因此,精氨酸和碳酸氢钠缓冲液比pbs更为优选。
(试验例6)
当使用探测器向sd大鼠(250~300g/只,5周龄)口服给药1ml的含有化学致癌性二甲基苯并蒽(dmba)10mg/ml的玉米油溶液时,在12~14周后发生乳腺癌。
由此,对于发生了乳腺癌的sd大鼠(1组5只),仅静脉内(i.v.)给药聚羟丙基甲基丙烯酰胺键合的吡柔比星(p-thp组),或者除i.v.给药p-thp之外,还涂布硝酸甘油(0.2mg/小鼠)(p-thp+ng组)。在试验期间,该给药共计进行四次。此外,p-thp的给药量均为5mg/kg。
在这些药物的给药后0天至140天之间,测定肿瘤体积(mm3)(图5)。其结果,与仅给药p-thp的组相比,除给药p-thp之外还涂布硝酸甘油的组,显示出显著更高的肿瘤抑制效应。
(试验例7)
将s-180小鼠肉瘤移植到ddy小鼠的腹膜腔内,每隔10天基于小鼠腹水进行传代得到的物质,移植到约106只6周龄的ddy小鼠的皮下,10天左右发生直径5~6mm的肿瘤。
由此,对于发生了s-180肉瘤的小鼠,仅静脉内(i.v.)给药p-thp(15mg)(p-thp组),或者除i.v.给药p-thp(15mg)之外,还涂布硝酸甘油(0.1mg/小鼠)(p-thp+ng组)。
在这些药物的给药后0天至40天之间,测定肿瘤体积(mm3)(图6)。其结果,与仅给药p-thp的组相比,除给药p-thp之外还涂布硝酸甘油的组,显示出显著更高的肿瘤抑制效应。
(试验例8)
将氧化偶氮甲烷(azm)(10mg/kg腹膜内(i.p.)给药)与硫酸葡聚糖钠(2%,0.2~1.0ml口服(p.o.)给药)给药1周,然后对于在第100天发生的小鼠的大肠肿瘤模型,i.v.给药15mg/kg的p-thp一次(p-thp组)、除i.v.给药p-thp(15mg)之外还涂布硝酸甘油软膏(0.1mg/小鼠)(p-thp+ng组)、除给药p-thp(15mg)之外还i.p.给药l-精氨酸(10~50mg/小鼠)(p-thp+arg组)、或者除给药p-thp(15mg)之外还i.p.给药羟基脲(hu)(1.5mg/小鼠)(p-thp+hu组)。
在这些药剂给药后,计算出大肠中所有肿瘤结节的各直径的总和(mm)(图7)。需要说明的是,这里提到的肿瘤结节的直径是指,将罗丹明标记的bsa(牛血清白蛋白)以1mg/小鼠地静脉注射,第二天将小鼠在尿烷麻醉下切除大肠,用卡尺测定通过ivis装置的肿瘤结节的荧光点的大小得到的值。
其结果为,与p-thp组相比,p-thp+ng组、p-thp+arg组和p-thp+hu组均显示出显著地高肿瘤抑制效应。
- 还没有人留言评论。精彩留言会获得点赞!