前列环素类似物制剂的制作方法
本发明涉及用以原位产生用于活性剂的控制释放的组合物的制剂前体(前体制剂)和用所述制剂进行治疗的方法。特定来说,本发明涉及两亲性组分和至少一种前列环素(prostacyclin)类似物的前体制剂,其在暴露于水性流体诸如体液后经历相变,由此形成控制释放组合物。
背景技术:
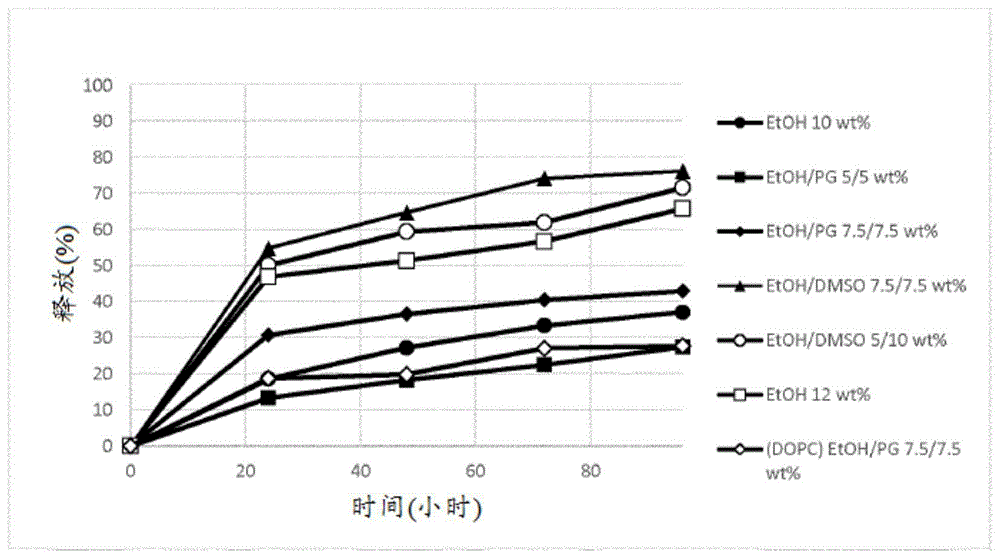
:包括药物、营养物、维生素等的许多生物活性剂具有“功能性窗口”。也就是说,存在可观察到这些试剂会提供某一生物作用所历经的一定浓度范围。当在身体的适当部分中的浓度(例如在局部中或如通过血清浓度所说明)下降至某一水平以下时,无有益作用可归因于试剂。类似地,通常存在在高于其时通过使浓度增加不会获得进一步益处的上部浓度水平。在一些情况下,使浓度增加高于特定水平会产生不合需要或甚至危险作用。一些生物活性剂具有长久生物半衰期和/或宽广功能性窗口,因此可不定期地施用,从而历经实质性时期(例如6小时至数天)维持功能性生物浓度。在其他情况下,清除率是较高的和/或功能性窗口是狭窄的,因此,为维持生物浓度在这个窗口内,需要以少量方式进行定期(或甚至连续)给药。这在非口服施用(例如胃肠外施用)途径是合乎需要的或必要的时可为特别困难的,因为自我施用可为困难的,因此导致不便和/或不良顺应性。在所述情况下,对于单次施用,将为有利的是历经需要活性所处的整个时期都在治疗水平下提供活性剂。一个特定类别的具有高清除率和短半衰期的活性剂是前列环素和它的类似物。前列环素是类花生酸(eicosanoid)家族的一个内源性成员,并且涉及于包括血小板活化、血管舒张和血压调控的若干过程中。当提及合成源性物质时,前列环素也称为依前列醇(epoprostenol),并且所述术语在本文中可互换使用。依前列醇在1995年由fda核准用于治疗肺动脉高血压(pah)。pah是特征在于平均肺动脉压(mpap)是≥25mmhg,伴有正常肺动脉楔压(pawp)(≤15mmhg)的潜在致命性疾患。然而,因为依前列醇自身具有小于1分钟的体内半衰期,所以它需要通常通过中心静脉导管来连续施用。用于静脉内疗法的依前列醇钠以(glaxosmithkline)进行销售。从2008年以来,室温稳定的依前列醇制剂(actelionpharmaceuticals)也已可用。据信在世界范围内估计有100,000至200,000名个体受pah的影响。已知具有较长半衰期的若干前列环素类似物,包括伊洛前列素(iloprost)(bayer)和曲前列环素(treprostinil)。曲前列环素在2002年由fda核准,并且具有2.9至4.6小时的血浆半衰期。尽管相较于依前列醇具有较长半衰期,但连续静脉内输注或定期皮下施用曲前列环素通常仍然是必要的。静脉内疗法需要手术插入中心静脉导管,携带感染和血栓形成风险,并且对于患者来说当然是不舒适的。依前列醇也可通过吸入或口服途径来施用。然而,相比于静脉内途径,这些途径提供依前列醇的较低累积剂量。因此,它们可不适于所有患者。(unitedtherapeuticscorporation)是一种被设计用于静脉内或连续皮下注射的曲前列环素制剂。连续皮下注射借助于微量输注泵来实现。尽管这解决与泵设备庞大相关的一些问题,但它仍然不理想,并且此外,推荐的是患者可快速获取备用输注泵。尽管定期皮下施用在某种程度上解决静脉内或连续皮下施用、口服或吸入途径的缺点,但施用部位疼痛是在大多数患者的情况下的重大障碍(由85%的患者所经受),并且导致几乎所有的归因于不利事件的曲前列环素治疗退出(长期研究群体中的总计23%)。到目前为止,这已在可能的程度上通过适当部位选择来处理。部位疼痛在部位变化之后前几天内达到峰值,并且在一些情况下,持续4周或更久使用单一部位可为有帮助的和安全的。存在对前列环素类似物的以下制剂的明显需要,所述制剂在储存时是稳定的,可在不需要通过中心静脉导管或通过连续皮下施用来连续施用下加以施用,不易经受机械故障的风险和/或可在较小频率下施用,同时相比于现有皮下制剂导致较小部位疼痛。本发明解决这些不足中的一些或全部。经受pah治疗的患者通常需要治疗剂量维持一段可观时期,并且通常需要持续许多个月或许多年进行治疗。因此,允许历经较长时期装载和控制释放较大剂量的储槽系统将提供超过常规递送系统的重大优势。就此而言,已开发含有曲前列环素的聚合物递送系统,诸如已经受1期临床试验的transcon曲前列环素(ascendispharma)。transcon曲前列环素被设计为曲前列环素的每日一次自我施用皮下注射液,并且基于聚合物递送系统,尤其是基于聚(噁唑啉)或peg的聚合物。transcon曲前列环素意图与连续输注的前列环素类似物提供相同功效,但采用更安全和更适宜的施用途径,伴有与当前胃肠外施用途径相关的部位反应和血流感染风险降低。通常用于降解缓慢释放制剂的聚乳酸、聚乙醇酸和聚乳酸共乙醇酸聚合物也是至少一些患者的某一刺激的原因。特定来说,这些聚合物通常含有某一比例的酸性杂质诸如乳酸和乙醇酸,其将在施用时刺激注射部位。当聚合物接着分解时,乳酸和乙醇酸是降解产物,以致导致进一步刺激。尽管由transcon曲前列环素在患者舒适性以及在某种程度上施用频率(每日一次)较小方面提供潜在优势,但即使使用不分解成酸性杂质的聚合物诸如peg,聚合物系统也倾向于具有高粘度,因此需要通过宽针来注射和/或仅提供相当短暂持续时间的产品。peg接枝于活性剂诸如曲前列环素通常使生物寿命增加,但可干扰结合,并且当前不能提供将在注射之间持续数天保持活性的产品。由于宽针施用和/或刺激性内含物的组合影响,所以在施用部位处的不适和结缔疤痕组织的形成常常在程度上大于合乎需要的程度。这在提出的曲前列环素制剂的情况下得以增加,因为注射是至少每日而非每周或更长周期进行的。因此,历经长久治疗持续时间,必须在少数部位处进行多次刺激性施用,或在利用许多部位的情况下,伴有所引起的广泛分布的受试者不适。显然,将为优势的是提供可易于通过窄针来施用,由此使在程序期间的患者不适降低以及导致较小部位疼痛的具有低粘度的系统,诸如均质溶液、精细粒子分散液或l2相。这个易于施用在患者将采用自我施用方案以及可已正在每天自我施用数次(如同采用若干现有曲前列环素治疗剂的情况)时是特别重要的。提供具有数天的持续时间,但施用足够复杂以致它需要由健康护理专业人员进行处理的持续制剂对于所有患者都将不是超过每日两次或每日进行自我施用的优势,并且可能更加昂贵。提供给予足够长久持续时间以使得有理由去健康专业人员处就诊以达成施用的制剂和/或可易于自我施用的制剂将是重大优势。降低在实际施用之前健康护理专业人员或患者的准备时间也是重要问题。从药物递送观点来看,聚合物储槽组合物通常也具有仅接受相对较低药物装载量,以及具有“爆发/迟滞”释放概况的缺点。尤其是当以溶液或前聚合物形式应用时,聚合基质的性质导致在最初施用组合物时的初始药物释放爆发。这继之以一段低释放时期,当基质的降解开始时,最终继之以释放速率增加以获得所需持续概况。这个爆发/迟滞释放概况可导致紧接在施用之后活性剂的体内浓度爆发超过功能性窗口,接着在迟滞时期期间回降至功能性窗口的底部,随后达到持续功能性浓度持续一段时期。显然,从功能性和毒理学观点来看,这个爆发/迟滞释放概况是不合需要的,并且可为危险的。由于在“峰值”点时不利作用具有危险性,所以这也可限制可提供的平衡浓度。此外,迟滞期的存在可需要在储槽治疗的启动时期期间以重复注射进行补充给药以在从储槽提供的活性剂的浓度处于亚功能性时维持治疗剂量。控制释放制剂通常由生物可相容聚合物以例如植入物或可注射珠粒形式产生。聚合物微球体制剂通常必须借助于通常具有20规格或更宽的大针来施用。由于通常是聚合物混悬液的所用聚合给药系统的性质,所以这是必要的。将为优势的是提供可易于通过窄针来施用,由此使在程序期间的患者不适降低的具有低粘度的系统,诸如均质溶液、精细粒子分散液或l2相。易于施用在患者将进行自我施用时是特别重要的,而且当健康护理专业人员进行施用时也使他们的负担降低。在某些现有储槽系统的情况下,plga微珠粒和混悬液的制造另外是重大困难。特定来说,因为珠粒是颗粒,所以它们通常不能加以无菌过滤,并且此外,因为plga共聚物在高温下熔融,所以它们不能加以热处理以达成无菌性。因此,必须无菌地进行复杂制造工艺。在生物可降解聚合物微球体的情况下的其他问题包括在注射之前的复原复杂以及储存稳定性有限,两者均归因于递送系统和/或活性剂的聚集和降解。已描述某些肽的基于脂质的缓慢释放组合物。举例来说,wo2006/131730公开glp-1及其类似物的一种脂质储槽系统。这是一种高度有效制剂,但可被包括在制剂中的活性剂的浓度受限于它的溶解性。显然,较高活性剂浓度允许有可能使储槽产品持续时间较长,产品维持较高全身性浓度,以及产品具有较小注射体积,所述因素全都在适当情况下具有重大优势。因此,将具有重大价值的是建立较高浓度的活性剂可被包括在基于脂质的储槽制剂中所采用的方式,以及鉴定活性剂和递送系统的从装载、稳定性、制造和/或控制释放的观点来看是特别有效的组合。本发明者现已确定通过提供处于低粘度相诸如分子溶液或l2(反胶束)相的包含至少一种中性单酰基脂质、二酰基脂质或三酰基脂质和/或生育酚、任选至少一种磷脂、至少一种生物可相容有机单醇溶剂和至少一种前列环素类似物或其盐的前体制剂,可产生解决已知曲前列环素制剂的许多不足的前体制剂,并且其可被施加以提供前列环素类似物的控制释放。通过以仔细选择的比率使用特定组分,可产生具有超越现有前列环素类似物制剂的性能的性质的组合,以及提供超过已知曲前列环素组合物诸如或transcon曲前列环素的优势的储槽制剂。特定来说,前体制剂显示高度有利的释放概况,易于制造,可加以无菌过滤,具有低粘度(允许通常通过窄针来达成容易和较小程度疼痛的施用),允许并入高水平的生物活性剂(因此潜在允许使用较小量的组合物和/或活性剂),需要浅层注射和/或在体内形成具有“低爆发”释放概况的所需非层状储槽组合物。组合物也由无毒、生物可耐受和生物可降解物质形成,所述组合物可通过单次肌肉内或皮下注射而非中心静脉导管或连续皮下注射来施用,并且适于自我施用。前体制剂可另外在注射时具有极低刺激水平,以及在优选情况下,在注射部位处不具有刺激(包括短暂刺激)。相比于同样提出的“缓慢释放”制剂,前体制剂可以较小频率施用,从而导致更好的患者顺应性和/或较小的归因于重复频繁施用的刺激。本发明制剂在施用之后产生非层状液晶相。在生物活性剂的递送中使用非层状相结构(诸如非层状液晶相)现时是相对广为接受的。一种最有效的脂质储槽系统描述于wo2005/117830中,并且在那个文件中描述一种高度优选的脂质储槽。然而,对于实现在若干方面具有改进性能的储槽制剂,仍然留有余地,并且特定来说,惊人改进可通过仔细选择和优化先前研究中公开的组分和比例的范围来实现。本发明组合物超过聚合物制剂诸如plga微球体的优势包括易于制造(包括灭菌)、处理和使用性质与活性剂的低初始释放(“低爆发概况”)组合。这可被定义为在一周给药时期的前24小时期间血浆浓度相对于时间曲线下面积是整个曲线的曲线下面积(从时间零点至无穷或从时间零点至末个取样时间点测量或外推)的小于50%,更优选小于40%,并且最优选小于30%。此外,它可被定义为在注射前体制剂之后活性剂的体内最大血浆浓度(cmax)是在治疗时期期间的平均血浆浓度(cave)的至多10倍,优选是至多8倍,并且最优选是至多5倍(即cmax/cave≤10,优选≤8,更优选≤5)。技术实现要素:本发明提供一种包含脂质赋形剂、有机醇溶剂和前列环素类似物以及某些任选组分的适当组合的药物制剂,其可作为储槽前体制剂(为简洁起见在本文中称为前体制剂)用于解决一种或多种上述需求。本发明者已确定通过优化这些组分,可产生具有高度有利的性质组合的尤其是曲前列环素的前列环素类似物的储槽组合物和相应前体制剂。在第一实施方案中,本发明提供一种前体制剂,其包含:a)单酰基脂质、二酰基脂质或三酰基脂质和/或生育酚中的至少一者;b)任选至少一种磷脂;c)至少一种生物可相容有机溶剂;和d)至少一种前列环素类似物或其盐;其中所述前体制剂在与过量水性流体接触后任选但优选形成或能够形成至少一种液晶相结构。在可适用于本发明的所有方面的一优选实施方案中,前列环素类似物含有3,4-顺式稠合环戊烷环、在所述环戊烷环的1位处的oh基团和在所述环戊烷环的2位处的c1-10基团,这些结构在本文中加以更详细定义。前列环素类似物可例如具有如本文指示的式i、ia、ib或ic。本发明的前列环素类似物将通常在分子内包括羧酸部分,或可为其盐。然而,当前列环素类似物不含有酸单元,以及不能够形成盐时,如本文所用的术语“游离酸”应解释为中性分子(例如中性酯)。在另一优选实施方案中,前列环素类似物(游离酸)具有小于500g/mol的分子量,并且不是多肽。在另一优选实施方案中,前列环素类似物(游离酸)在前体制剂的0.1至10%,优选在0.2至6%的水平下存在。在一实施方案中,前列环素类似物(游离酸)在诸如0.2至5%、0.5至5%,尤其是0.2至4%或0.75至4%的水平下存在。在另一优选实施方案中,前列环素类似物包含以下或由以下组成:曲前列环素(tpn)或其盐,优选是曲前列环素钠盐。在一优选实施方案中,组分c)包含至少一种选自由以下组成的组的溶剂或由至少一种选自由以下组成的组的溶剂组成:醇、胺、酰胺、亚砜和/或酯。在一优选实施方案中,c)包含以下或由以下组成:乙醇或乙醇和丙二醇的混合物,优选地,其中乙醇与pg的比率是1:1至10:1,更优选是1.5:1至8:1,最优选是2:1至5:1(例如约3:1)。在另一优选实施方案中,如本文所定义,在25℃和60%rh下,在3个月之后,优选在6个月之后,尤其在12个月之后,就如通过hplc测量的活性剂测定而言,前体制剂具有至少96%,优选至少97%,尤其至少98%的稳定性。在另一优选实施方案中,在40℃和75%rh下,在1个月之后,优选在3个月之后,尤其在6个月之后,就如在储存之后通过hplc测量的活性剂测定而言,前体制剂具有至少96%,优选至少97%,尤其至少98%的稳定性。在一尤其优选实施方案中,组分a)包含gdo或由gdo组成,组分b)包含大豆pc或由大豆pc组成;组分c)包含乙醇,并且任选包含丙二醇;并且组分d)包含以下或由以下组成:曲前列环素或其盐(例如钠盐)。在第二方面,本发明涉及如本文定义的前体制剂用以持续施用前列环素类似物的用途。在另一方面,本发明提供用作药剂(例如用于治疗本文所述的疾患)的根据第一实施方案的前体制剂或通过使所述前体制剂暴露于过量水性流体来获得的组合物。在另一方面,本发明提供一种用于治疗人或非人哺乳动物受试者的方法,其包括向所述受试者施用如本文定义的前体制剂。在一个实施方案中,治疗方法(以及相应用途和其他方面)是一种用于治疗人或非人哺乳动物受试者(尤其是有需要者)的方法。在另一实施方案中,治疗方法(以及相应用途和其他方面)是一种用于治疗至少一种选自以下的疾患的方法:肺动脉高血压(pah)、pah相关慢性阻塞性肺病(copd)、重度雷诺氏病(raynaud’sdisease)、缺血和相关疾患。在一实施方案中,治疗方法涉及每1至60天,优选每1、2、3、7、14、21、28、30或60天(例如±3天或在任何情况下±20%),最优选每7(±1)天或每14(±2)天或每30(±3)天施用如本文定义的前体制剂。在一实施方案中,治疗方法涉及在0.005至2.5mg/kg/周的水平下,优选在0.01至1mg/kg/周,尤其在0.015至0.7mg/kg/周的水平下施用所述前列环素类似物或其盐。在另一方面,本发明涉及一种如本文所述的前体制剂,其用于如本文所述的治疗方法中(包括在本文所述的所有疾病、疾患、剂量、方法或施用和施用方案的情况下使用)。在另一方面,本发明涉及如本文定义的前体制剂用以制造用于在体内形成储槽的药剂的用途,所述储槽用于治疗至少一种选自以下的疾患:肺动脉高血压(pah)、pah相关copd、雷诺氏病、缺血和相关疾患。在另一方面,本发明提供一种含有如本文定义的前体制剂的预填充施用装置。在另一方面,本发明涉及一种药盒,其包括如本文定义的施用装置,优选包括自动注射器、药筒和/或笔。附图说明图1.从表1选择的制剂的随时间(a)和时间的平方根(b)变化的体外释放概况。图2.在使用制剂b1和b2进行试探性研究给药期间大鼠的体重变化的结果(参见实施例2和表2)。图3.所选制剂l-aa的粘度(参见实施例3和表6)图4.制剂n、p、q、r和s的体外释放概况(累积释放百分比)(a),其中0-20%释放区域处于扩展视图(b)中。图5.在水性介质中平衡之后,制剂l-s在25℃、37℃和42℃下的x射线衍射图。图6.在水性介质中平衡之后,制剂t-aa在25℃、37℃和42℃下的x射线衍射图。图7.在施用制剂ee、ff、gg或hh之后大鼠中tpn的平均血浆浓度。图8.制剂ff、ee、x和hh的体外释放概况(累积释放百分比)。图9.在将含3、15、22.5和30mgtpn的前体制剂单次皮下注射至雄性和雌性比格犬中之后的平均曲前列环素血浆浓度-时间概况。图10.在将含3、15、22.5和30mgtpn的前体制剂单次皮下注射至雄性和雌性比格犬中之后的平均曲前列环素auc0-168h值。具体实施方式本发明制剂在施用之后产生非层状液晶相。在生物活性剂的递送中使用非层状相结构(诸如液晶相)现时是相对广为接受的。一种最有效的通用脂质储槽系统描述于wo2005/117830中,并且在那个文件中概括地描述一种适于在本发明中使用的脂质基质,所述专利的全部公开内容据此以引用的方式并入本文。关于对所述制剂的最有利相结构的描述,应注意wo2005/117830中的讨论,并且特别应注意其第29页。除非另外指示,否则遍及本文所有%都以重量指定。此外,当上下文允许时,指示的重量%是包括本文指示的所有组分的总前体制剂的%。不管使用酸还是其盐,前列环素类似物的重量百分比都将基于游离酸的重量加以计算。前体制剂可任选基本上仅由本文指示的组分(当适当时包括本文在以下以及随附权利要求中指示的额外任选组分)组成,并且在一个方面,完全由所述组分组成。当制剂被指示为“基本上由某些本文组分组成”时,此时指定组分提供那个制剂的必需性质,诸如此时指定组分占制剂的至少95%,优选是至少98%。优选地,本发明的前体制剂是分子溶液或具有l2相结构(在施用之前)。在施用之后,前体制剂形成非层状(例如液晶)相。这种相变通常由从生理环境吸收水性流体所导致,如本文所指示。尽管先前已在wo2012/160213中确定倘若存在单醇溶剂,那么仔细控制量的水可被容忍,但应了解在施用后,前体制剂暴露于大量水性流体。通常,在与至少等体积量的水性流体接触后,前体制剂将形成非层状相。本发明者现已惊人地确定通过适当选择脂质组分以及前列环素类似物和生物可相容有机溶剂的类型、绝对量和比率,可致使由本发明的前体制剂形成的储槽组合物的释放性质高度有利,并且优于现有曲前列环素储槽制剂。特定来说,单次施用前列环素类似物的释放持续时间如果远超现有曲前列环素储槽的释放持续时间,那么体内最大血浆浓度仅是在给药时期期间的平均浓度或甚至最小浓度的较小倍数。组分a)-酰基脂质/生育酚组分a)的优选范围是15-85wt%,优选是20-80%,优选是30-60wt%,优选是35-55%,诸如是38-52%,尤其是38至52%。在一些实施方案中,约43%(例如41至45%)的水平是特别优选的。组分b)的优选范围是15-85wt%,优选是20-80%,优选是30-60wt%,优选是35-55%,诸如是38-52%,尤其是38至52%。在一些实施方案中,约43%(例如41至45%)的水平是特别优选的。a:b的比率通常是40:60至60:40,优选是45:55至55:45,并且更优选是47:53至53:47。约50:50(例如±2)的比率是高度有效的。如本文指示的组分“a”包含单酰基脂质或二酰基脂质和/或生育酚中的一者或多者。最优选地,组分a)包含单酰基脂质或二酰基脂质或由单酰基脂质或二酰基脂质组成,因此具有一个或两个非极性“尾部”基团。用于本发明中的酰基甘油(例如单酰基甘油或二酰基甘油)作为纯化合物在25℃下在水中将通常不形成非层状液晶相结构。在一个实施方案中,组分a)可为单酰基脂质。单酰基脂质含有极性“头部”基团和一个非极性“尾部基团”。“头部”基团可为甘油、二甘油、糖部分(诸如基于肌醇和葡萄糖基的部分);以及多元醇的酯,诸如乙酸酯或丁二酸酯。一个优选类别的单酰基脂质是诸如脱水山梨糖醇的己糖醇酐的酯。在这个术语中,“己糖醇酐”表示式hoch2(choh)4ch2oh己糖醇,其已由于损失一当量的水而环化以形成五元环或六元环,优选是五元呋喃糖环。脱水山梨糖醇是一特别优选的头部基团。头部基团优选通过酯键来连接于尾部基团。在以下讨论适合尾部基团。在一特别优选实施方案中,组分a)包含以下或由以下组成:至少一种二酰基脂质,优选是二酰基甘油(dag)。二酰基脂质包含如上所述的极性头部基团和优选通过酯键来连接于所述极性头部基团的两个无极性尾部基团。二酰基脂质的最优选极性头部基团是甘油。非极性基团可具有相同或不同数目的碳原子,并且可各自独立地是饱和的或不饱和的。非极性基团的实例包括c6-c32烷基和烯基,其通常以长链羧酸的酯形式存在。这些常常通过提及碳链中的碳原子数目和不饱和度数目来描述。因此,cx:z指示具有x个碳原子和z个不饱和度的烃链。实例特别包括月桂酰基(c12:0)、肉豆蔻酰基(c14:0)、棕榈酰基(c16:0)、植烷酰基(c16:0)、棕榈油酰基(c16:1)、硬脂酰基(c18:0)、异硬脂酰基(c18:0)、油酰基(c18:1)、反油酰基(c18:1)、亚油酰基(c18:2)、亚麻酰基(c18:3)、花生四烯酰基(c20:4)、山嵛酰基(c22:0)和二十四酰基(c24:9)。因此,典型非极性链基于天然酯脂质的脂肪酸,包括己酸、辛酸、癸酸、月桂酸、肉豆蔻酸、棕榈酸、植烷酸、棕榈炔酸、硬脂酸、油酸、反油酸、亚油酸、亚麻酸、花生四烯酸、山嵛酸或二十四酸,或相应醇。优选非极性链是棕榈酸、硬脂酸、油酸和亚油酸,特别是油酸。任何数目的单酰基脂质或二酰基脂质的混合物都可用作组分a)。优选地,这个组分将包括至少一部分的c18脂质(例如具有一个或多个(即一个或两个)c18:0、c18:1、c18:2或c18:3非极性基团的dag),诸如脱水山梨糖醇单油酸酯(smo)、甘油二油酸酯(gdo)和/或甘油二亚油酸酯(gdl)。一高度优选实例是包含至少50%,优选至少80%,并且甚至包含大致上100%gdo的dag。因为gdo以及其他单酰基甘油和二酰基甘油是源于天然来源的产品,所以通常存在某一比例的具有其他链长的“污染”脂质等。在一个方面,如本文所用的gdo因此用于指示具有相伴杂质的任何商业等级gdo(即商业纯度gdo)。这些杂质可通过纯化来分离和移除,但倘若等级是一致的,那么这很少是必要的。然而,如果必要,那么“gdo”可为基本上化学纯净gdo,诸如是至少80%纯净,优选是至少85%纯净,并且更优选是至少90%纯净gdo。用作组分a)的全部或一部分的一个替代性或额外优选类别的化合物是生育酚。如本文所用,术语“生育酚”用于指示非离子脂质生育酚,常常称为维生素e,和/或其任何适合盐和/或类似物。适合类似物将是在暴露于水性流体后提供相特性、无毒性和相变的那些,所述特征是本发明组合物的特征。所述类似物作为纯化合物在25℃下在水中将通常不形成非层状液晶相结构。最优选的生育酚是具有以下结构的生育酚自身。显然,特别是当这从天然来源纯化时,可存在小比例的非生育酚“污染物”,但此将不足以改变有利相特性或无毒性。通常,以重量计,生育酚将含有至多10%的非生育酚类似物化合物,优选含有至多5%,并且最优选含有至多2%。生育酚在本发明的一个实施方案中,组分a)基本上由生育酚,特别是如上所示的生育酚组成。组分a)的一优选成分组合是至少一种二酰基甘油(dag-例如至少一种c16至c18dag,诸如gdo)与至少一种生育酚的混合物。以重量计,所述混合物包括2:98至98:2生育酚:dag,例如10:90至90:10生育酚:dag,并且尤其包括20:80至80:20的这些化合物。生育酚与其他酰基甘油诸如本文讨论的那些中的任一者的类似混合物也是适合的。组分b)-磷脂本发明的优选脂质基质中的任选组分“b”是至少一种磷脂。如同组分a)一样,这个组分包含极性头部基团和至少一个非极性尾部基团。组分a)与b)之间的差异主要在于极性基团。因此,非极性部分可适合地源于以上对于组分a)所考虑的脂肪酸或相应醇。磷脂(例如pc)将含有两个非极性基团。再次,c18基团是优选的,并且可与任何其他适合非极性基团特别是c16基团组合。用于本发明中的磷脂可为作为纯化合物在25℃下在水中不形成非层状液晶相结构的那些。或者,用于本发明中的磷脂可为在25℃下在水中形成非层状液晶相结构例如六方液晶相的那些。磷脂部分,甚至比任何二酰基甘油部分更适合,可源于天然来源。适合磷脂来源包括卵、心脏(例如牛心脏)、脑、肝(例如牛肝)以及包括大豆的植物来源。所述来源可提供可包含任何磷脂混合物的组分b的一种或多种成分。适于组分b)的极性头部基团包括磷脂酰胆碱、磷脂酰乙醇胺、磷脂酰丝氨酸和磷脂酰肌醇。最优选的是磷脂酰胆碱(pc)和/或磷脂酰乙醇胺(pe)。已在wo2013/038460和wo2013/083459中显示使用磷脂总量的至少50重量%的pe可导致储槽稳健性改进。从wo2016/066655得知基于三酰基脂质的脂质缓慢释放基质在暴露于水性流体时在不需要磷脂组分存在下可形成储槽组合物,但磷脂也可存在。因此,在一个实施方案中,组分a)包含三酰基脂质,由三酰基脂质组成或基本上由三酰基脂质组成,并且组分b)是任选的。然而,在另一实施方案中,如果组分a)包括大于50%单酰基脂质或二酰基脂质、或至少一种生育酚、或任何这些组分的混合物,那么磷脂组分b)将优选存在。在一个实施方案中,组分a)包括小于50%(例如0至49%或0.1至45%)三酰基脂质(基于组分a)的总量),并且组分b)存在(例如在前体制剂的20至80wt%下或在如本文各种实施方案中指示的其他量下)。在本发明中,特别优选的是组分b)包含一种或多种pc或由一种或多种pc组成。举例来说,组分b)的至少50%的头部基团应是pc,优选是超过65%的头部基团,尤其是超过85%或超过90%。可使用来自这些或其他来源的任何单一pc或pc混合物,但包含大豆pc或卵pc的混合物是高度适合的。pc组分优选含有至少50%大豆pc或卵pc,更优选含有至少75%大豆pc或卵pc,并且最优选含有基本上纯净的大豆pc或卵pc。在一个可适用于本发明的所有方面的实施方案中,组分b)包含pc或由pc组成。优选地,pc源于大豆。优选地,pc包含18:2脂肪酸作为主要脂肪酸组分,伴有16:0和/或18:1脂肪酸作为次要脂肪酸组分。这些优选在1.5:1与6:1之间的比率下存在于pc中。具有约60-65%18:2、10至20%16:0、5-15%18:1,其余部分主要是其他16碳和18碳脂肪酸的pc是优选的,并且代表大豆pc。在也可适用于本发明的所有方面的一替代性但同等优选实施方案中,pc组分可包含合成二油酰基pc(dopc)。据信这提供增加的稳定性,因此对于需要稳定长期储存和/或在体内具有长久释放时期的组合物将是特别优选的。在这个实施方案中,pc组分优选含有至少50%合成二油酰基pc,更优选含有至少75%合成二油酰基pc,并且最优选含有基本上纯净的合成二油酰基pc。任何其余pc都优选是如上大豆pc或卵pc。在一个实施方案中,本发明的前体制剂至少部分地包含合成dopc(即具有至少95%pc头部基团和至少90%油酰基(c18:1)酰基的pc),并且在至少6个月,更优选至少12个月,并且最优选至少24个月之后,具有在15-25℃下的储存稳定性,其被定义为小于5%活性剂降解,如通过hplc所测定。因为本发明的前体制剂将向受试者施用以达成控制释放前列环素类似物,所以重要的是各组分是生物可相容的。就此而言,用于本发明的前体制剂中的优选脂质基质是高度有利的,因为pc与dag两者均是良好耐受的,并且在体内分解成天然存在于哺乳动物身体中的组分。合成或高度纯化pc诸如二油酰基磷脂酰胆碱(dopc)和棕榈酰基油酰基磷脂酰胆碱(popc)以及本文所述的其他各种高纯度pc作为组分b)的全部或一部分是高度适当的。在一高度优选实施方案中,组分b)是由具有包含至少95%磷脂酰胆碱的极性头部基团和各自独立地具有16至20个碳的两个酰基链的磷脂组成的“高纯度”pc,其中至少一个酰基链在碳链中具有至少一个不饱和度,并且在两个碳链内存在至多四个不饱和度。通常,这可为具有至少95%pc头部基团和至少95%具有0至3个不饱和度的c16至c20酰基链的pc。合成二油酰基pc最优选是1,2-二油酰基-sn-甘油基-3-磷酸胆碱,并且其他合成pc组分包括ddpc(1,2-二癸酰基-sn-甘油基-3-磷酸胆碱);depc(1,2-二芥酰基-sn-甘油基-3-磷酸胆碱);dlopc(1,2-二亚油酰基-sn-甘油基-3-磷酸胆碱);dlpc(1,2-二月桂酰基-sn-甘油基-3-磷酸胆碱);dmpc(1,2-二肉豆蔻酰基-sn-甘油基-3-磷酸胆碱);dopc(1,2-二油酰基-sn-甘油基-3-磷酸胆碱);dppc(1,2-二棕榈酰基-sn-甘油基-3-磷酸胆碱);dspc(1,2-二硬脂酰基-sn-甘油基-3-磷酸胆碱);mppc(1-肉豆蔻酰基-2-棕榈酰基-sn-甘油基3-磷酸胆碱);mspc(1-肉豆蔻酰基-2-硬脂酰基-sn-甘油基-3–磷酸胆碱);pmpc(1-棕榈酰基-2-肉豆蔻酰基-sn-甘油基-3–磷酸胆碱);popc(1-棕榈酰基-2-油酰基-sn-甘油基-3-磷酸胆碱);pspc(1-棕榈酰基-2-硬脂酰基-sn-甘油基-3–磷酸胆碱);smpc(1-硬脂酰基-2-肉豆蔻酰基-sn-甘油基-3–磷酸胆碱);sopc(1-硬脂酰基-2-油酰基-sn-甘油基-3-磷酸胆碱);和sppc(1-硬脂酰基-2-棕榈酰基-sn-甘油基-3-磷酸胆碱)或其任何组合。组分a)和b)的一特别有利组合是smo与pc,gdo与pc,尤其是gdo与大豆pc和/或“高纯度”pc。各组分的适用于组合的适当量是本文对于任何组合中的个别组分指示的那些量。当上下文允许时,这也适用于本文指示的任何组分组合。组分a:b的比率在40:60至60:40的范围内。优选地,a:b比率在45:55至55:45,更优选在47:53至53:47的范围内。最优选地,a:b比率是约50:50,诸如48:52至52:48。在一个实施方案中,组分a)的绝对量将是40至47%,组分b)的绝对量将是40至47%,a:b的比率将是48:52至52:48,组分c)的量将是5至20%,优选是8至12%,其中组分c)由在2.5:1至3.5:1的比率下的乙醇和丙二醇组成,并且组分d)将是在2.5至50mg/ml(基于游离酸),诸如在5至50mg/ml(基于游离酸)下的曲前列环素钠。组分c)–生物可相容有机溶剂本发明的前体制剂的组分c)是生物可相容有机溶剂。因为前体制剂将在施用(例如在体内)之后通常在与水性流体接触后产生储槽组合物,所以合乎需要的是这个溶剂可为受试者所耐受,并且能够与水性流体混合,和/或扩散或溶解出前体制剂进入水性流体中。因此,具有至少中度水溶性的溶剂是优选的。组分c)包含选自由以下组成的组的生物可相容有机溶剂或由选自由以下组成的组的生物可相容有机溶剂组成:包括单醇溶剂以及二醇溶剂和多元醇溶剂的醇、胺、酰胺、亚砜或酯。特别优选的是组分c)包含单醇溶剂或由单醇溶剂组成。组分c)可包含两种或更多种来自以上溶剂的清单的组分,特别是单醇溶剂和选自酰胺、亚砜或二醇溶剂的溶剂。不是单醇溶剂的任何溶剂都可在本文中称为共溶剂。当存在两种或更多种溶剂时,尤其优选的组合是乙醇和酰胺(诸如乙醇和nmp)、乙醇和亚砜(诸如乙醇和dmso)、或乙醇和二醇溶剂或多元醇溶剂(诸如乙醇和pg)。一高度优选的溶剂组合是乙醇和pg,特别是当乙醇与pg的比率是1:5至20:1,优选是1:1至10:1,更优选是1.5:1至8:1,最优选是2:1至5:1(例如约3:1,诸如2.8:1至3.2:1)时。组分c)可包含以下或由以下组成:乙醇、丙醇、异丙醇、苯甲醇或其混合物。最优选地,组分c)包含乙醇或由乙醇组成。前体制剂中组分c)的量将对若干特征具有可观影响。特定来说,粘度和释放速率(和持续时间)将随溶剂水平而显著改变。因此,溶剂的量将至少足以提供低粘度混合物,但将另外加以确定以便提供所需释放速率。这可鉴于以下实施例通过常规方法来确定。通常,1至30%,特别是2至25%的溶剂水平将提供适合释放和粘度性质。这将优选是2至20%,优选是5至15%,并且约10%(例如10±3%)的量是高度有效的。这些水平包括作为组分c)的一部分存在的如上提及的任何共溶剂。如上所指示,本发明的前体制剂中组分c)的量将至少足以提供组分a)、b)、c)和d)的低粘度混合物(例如分子溶液),并且将易于通过标准方法来确定用于任何特定组分组合的组分c)的量。相特性可通过诸如目视观察与偏光显微术、x射线散射和衍射技术、核磁共振、以及低温透射电子显微术(低温tem)组合的技术来分析以找寻溶液、l2或l3相、或液晶相,或如在低温tem的情况下找寻所述相的分散片段。粘度可通过标准手段直接测量。如上所述,适当实用粘度是可达成有效注射,并且特别是可达成无菌过滤的粘度。这将易于如本文所指示加以评估。组分a)、b)和c)的一高度优选组合是gdo、大豆pc和/或“高纯度pc”、和乙醇,或smo、大豆pc和乙醇。其他优选组合包括gdo/spc/乙醇/dmso、gdo/spc/乙醇/nmp和gdo/spc/乙醇/pg。如上所指示,各组分的适用于组合的适当量是本文对于任何组合中的个别组分指示的那些量。如本文所用的组分c)可为单一溶剂或适合溶剂的混合物,但将通常具有低粘度。这是重要的,因为本发明的一个关键方面是它提供具有低粘度的前体制剂,并且适合溶剂的主要作用在于使这个粘度降低。这个降低将是溶剂的较低粘度的影响和溶剂与脂质组合物之间的分子相互作用的影响的组合。本发明者的一个观察结果是本文所述的低粘度含氧溶剂与组合物的脂质部分具有高度有利和出乎意料的分子相互作用,由此在添加小体积的溶剂的情况下提供非线性粘度降低。“低粘度”溶剂组分c)(单一溶剂或混合物)的粘度应通常是在20℃下至多18mpas。这优选地是在20℃下至多15mpas,更优选是在20℃下至多10mpas,并且最优选是在20℃下至多7mpas。已在wo2012/160213中确定与“极性溶剂”或“共溶剂”诸如二醇或水组合使用醇溶剂允许某些基于脂质的控制释放组合物的性能显著改进。特定来说,已观察到添加二醇(诸如丙二醇)或水允许醇的比例增加而不不利地影响释放概况,和/或允许释放概况改进和/或允许活性剂的装载量较高。典型共溶剂将具有对应于它们的高极性的相比较来说较高介电常数。因此,适合共溶剂将通常具有在25℃下至少28,更优选是在25℃下至少30的介电常数。高度适合的实例包括水(约80)、丙二醇(约32)、二甲亚砜(约47)和n-甲基-2-吡咯烷酮(nmp,约32)。在一些实施方案中,组分c)的一特别适当的溶剂组合包括单醇溶剂和选自由以下组成的组的共溶剂:酰胺、亚砜或二醇。一尤其优选组合是乙醇和酰胺、乙醇和亚砜、或乙醇和二醇。特别优选组合是乙醇和丙二醇(pg);乙醇和二甲亚砜(dmso);以及乙醇和n-甲基-吡咯烷酮(nmp)。当存在时,共溶剂的适合量将通常是前体制剂的大于1重量%,例如2-15%,特别是4-12.%,尤其是4-10wt%。作为组分c)的单醇溶剂和共溶剂的组合在本发明组合物中具有潜在优势。特定来说,通过包括可与单醇组分混溶的某一共溶剂,可在注射部位处由醇内含物引起的轻微感觉可得以大致上消除。因此,在一个实施方案中,单醇组分:共溶剂的比率可在30:70至90:10,更优选在50:50至80:20,尤其在60:40至80:20的范围内。组分的量(w/w)近似相等是高度适当的。组分d)–前列环素类似物本发明的前体制剂含有至少一种前列环素类似物或其盐。以下显示前列环素和合成类似物贝前列素(beraprost)、依前列醇、伊洛前列素和曲前列环素。对前列环素类似物的选择对于本发明来说不是特别关键的,只要它实现所需治疗作用,并且不不利地影响前体制剂的相特性即可。然而,通常,前列环素类似物组分d)将具有一个或多个以下特征。首先,它优选是合成非肽。其次,它优选具有低于500amu,优选低于400amu的分子量(游离酸)。第三,它优选包含具有1-羟基取代基、在2位处的c3-12烷基、烯基或炔基、以及3,4-顺式稠合5元或6元环的环戊烷单元。参照以上给出的结构,将易于了解所用编号。第四,前列环素类似物优选包含羧酸和/或酯单元。如可由贝前列素、依前列醇、伊洛前列素和曲前列环素的结构所见,在前列环素与已知合成类似物贝前列素、伊洛前列素和曲前列环素之间共有这些结构特征。用于本发明的所有方面的前列环素类似物可特别包括式(i)前列环素类似物:其中:n是1或2;x是o、ch2、chf或cf2;r是h、r10,或通过连接单元来连接于聚乙二醇(peg);r1是h、f或c1-c10取代或未取代的烷基、烯基或炔基;r2’是h、f或c1-c6取代或未取代的烷基、烯基或炔基;r2是饱和或不饱和c1-12取代或未取代的烷基、烯基或炔基,优选是饱和或不饱和c1-10基团;r5是x(ch2)aco2r9,其中x是o或ch2,a是0至4,优选是1或2,并且其中r9是h、c1-c6取代或未取代的烷基、烯基或炔基或生物学上可接受的阳离子;r8和r8’各自独立地是h、f或c1-c6烷基、烯基或炔基,优选是h;n是1或2;并且:所有r4和r4’基团都各自独立地是h、f或c1-c6取代或未取代的烷基、烯基或炔基;并且r5’是h、f或c1-c6取代或未取代的烷基、烯基或炔基,优选是h;或:r5’和相邻r4和/或r4’基团形成5元、6元或7元取代或未取代的环,优选是6元环,并且最优选是取代或未取代的6元芳族环;并且任何额外r4和/或r4’基团都各自独立地是h、f或c1-c6取代或未取代的烷基、烯基或炔基。r10是诸如保护性部分或前药部分的基团。适合保护性部分和/或前药部分包括酯,包括在后续章节中定义的那些。在一优选实施方案中,如果n是1,那么结构具有式(i-a)其中r4和r4’各自独立地是h、f或c1-c6烷基,优选是h;并且r5’是h、f或c1-c6烷基,优选是h;其中其余取代基如上对于式(i)所定义;或具有结构式(i-b)其中取代基如上对于式(i)所定义;在另一优选实施方案中,如果n是2,那么结构具有式(i-c)其中r4和r4’各自独立地是h、f或c1-c6烷基,优选是h;其中其余取代基如上对于式(i)所定义。以下是特别优选实施方案。对于式(i)、(ia)、(ib)和(ic)中的各者:x优选是o或ch2;r1优选是h;r2优选包括连接于来自环戊烷单元的r2的第三碳原子的oh基团;r2’优选是h;r4和r4’全都优选是h或与r5’形成酚环;r8与r8’均优选是h;除以上优选实施方案之外,对于式(ia):r2优选是不饱和c6-c12基团,优选是不饱和c8-c10基团,尤其包括连接于来自环戊烷单元的r2的第三碳原子的oh基团。还更优选地,r2是甚至更优选地,r2是:或r2是:或r2是:甚至更优选地,r2是:或r2是:r5’优选是h;x是o或ch2;r5优选是ch2ch2ch2co2r9,其中r9如上所定义,r5尤其是ch2ch2ch2co2h;r优选是h。在一实施方案中,前列环素类似物可为伊洛前列素。对于式(ib):r2优选是不饱和c6-c12基团,优选是不饱和c8-c10基团,尤其包括连接于来自环戊烷单元的r2的第三碳原子的oh基团。还更优选地,r2是甚至更优选地,r2是:x优选是o;r5优选是ch2ch2ch2co2r9,其中r9如上所定义,r5尤其是ch2ch2ch2co2h;r优选是h。在一实施方案中,前列环素类似物可为贝前列素。对于式(ic):x优选是ch2;r优选是h,或通过连接基团来连接于peg;r2优选是饱和c6-c10基团,优选是饱和c8基团,尤其包括连接于来自环戊烷单元的r2的第三碳原子的oh基团。还更优选地,r2是:或r2是:甚至更优选地,r2是:或r2是:r5优选是och2co2r9,其中r9如上所定义,r5最优选是och2co2h。在一实施方案中,前列环素类似物可为曲前列环素。在任何本文指示的结构式中,当存在oh基团(尤其是作为基团or或作为基团r2的要素)时,这种基团可以前药形式加以保护。所述前药通常在体内水解以使游离–oh部分再生,并且可包括酯和/或缩醛基团。特别适合的前药如整体并入本文的us9394227中对于r1和r2位置所述。在所述情况下,当oh部分以前药形式加以保护时,任何–oh基团都可独立地以–o-r10基团形式加以保护,其中r10在各次出现时都独立地是以下中的一者:r11在各次出现时都独立地是烷基、烷基芳基、环烷基、杂环基、芳基或杂芳基,其各自可任选被取代;r12和r13在各次出现时都独立地是氢、c1-c6烷基或c3-c6环烷基,或r12和r13以及它们所连接的碳原子形成c3-c6环烷基环;r14在各次出现时都独立地是氢、r11、-c(═o)r11、-c(═o)or11或-c(═o)nr15r16;或r14和r12或r13连同它们所连接的原子一起形成杂环;r15和r16在各次出现时都独立地是氢、烷基、-烷基芳基、环烷基、杂环基、芳基或杂芳基;或r15和r16以及它们所连接的氮原子形成杂环或杂芳基环;j在各次出现时都独立地是0至4的整数;并且m在各次出现时都独立地是1至10的整数。在某些实施方案中,r11在各次出现时都独立地是甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基或叔丁基;r12和r13在各次出现时都独立地是氢、甲基、乙基、丙基或异丙基;r14在各次出现时都独立地是氢或r11;j是0;并且m是1。当对–oh基团进行取代时,这可独立地在任一–oh基团处,或优选将在所有游离–oh基团处。在一个优选实施方案中,作为or或作为基团r2的一部分存在的所有–oh基团都将以相同前药–o-r10基团加以保护。在一些实施方案中,存在的任一或各–oh基团都被取代成酯。可用于对“-oh”基团进行取代以提供前药的一些特定实例酯包括乙酯、异丙酯或丁二酸酯。在其中前列环素类似物d)是前药的所有实施方案中,优选的是所述前药被配制在包含以下的前体制剂中:a)单酰基脂质、二酰基脂质或三酰基脂质和/或生育酚中的至少一者;b)任选至少一种磷脂;和c)至少一种生物可相容有机溶剂;以及并且其中所述前体制剂在与过量水性流体接触后形成或能够形成至少一种液晶相结构。特定来说,在其中前列环素类似物d)是前药的所有实施方案中,优选的是所述前药被配制在包含以下的前体制剂中:a)二酰基脂质,最优选是甘油二油酸酯(gdo);b)至少一种磷脂,优选是磷脂酰胆碱(pc);和c)至少一种生物可相容有机溶剂;以及并且其中所述前体制剂在与过量水性流体接触后形成或能够形成至少一种液晶相结构。在所有实施方案中,根据式(i)的环系统的几何结构优选是:在一优选实施方案中,前列环素类似物具有小于500g/mol的分子量。最优选地,组分d)包含以下或由以下组成:贝前列素、依前列醇、伊洛前列素或曲前列环素(例如依前列醇、伊洛前列素或曲前列环素),最优选是曲前列环素。也可使用前列环素类似物的任何生物学上可接受的盐。当组分d)的量以重量百分比给出时,除非上下文另外允许,否则意指的是基于游离酸的重量。在一特别优选实施方案中,组分d)包含以下或由以下组成:曲前列环素游离酸(tpn)或其盐,最优选是曲前列环素钠盐(tpn(na))。也设想使用酯衍生物诸如乙酯或其他生物学上可容许的醇(包括二醇或多羟基醇诸如丙二醇或甘油)的酯,并且此可提供可适用于控制前列环素类似物的释放和/或它的生物半衰期的“前药”效应。本文指示任选使用“连接单元”或“接头”,特别是在式(i)的“r”位置或其他结构上的相应位点处。这种接头可例如在位置r处形成酯键,并且可借助于其中任一者都可根据需要加以取代或未取代的直链、支链和/或环状烷基和/或烯基部分、酯基团、酰胺基团、胺基团、醚基团、硫醇、硫酯和环状部分诸如吡咯烷和吡咯烷二酮(例如3-硫烷基-2,5-吡咯烷二酮)基团来接合较大部分诸如肽、蛋白质、peg基团。适用于本发明中的其他前列环素类似物包括所有前列环素受体激动剂诸如色利西帕(selexipag)。基于前列环素类似物游离酸,组分d)以0.1至15%,优选以0.1至10%,诸如以1至12%,尤其以2至8%的量存在。在一些实施方案中,前列环素类似物的水平可为4至8%。这些水平特别适于曲前列环素。本发明者的一惊人发现是前列环素类似物诸如曲前列环素的释放持续时间强烈取决于活性剂的量与溶剂组分c)的性质两者。因此,储槽的释放性质可通过改变这些参数中的一者或多者来调谐。此外,本发明者的一惊人发现是前列环素类似物诸如曲前列环素的释放可通过适当选择配制溶剂和溶剂比率来有效控制。一般来说,在单酰基脂质和/或二酰基脂质(诸如gdo)连同磷脂诸如磷脂酰胆碱一起的储槽制剂的情况下,活性剂的释放主要由制剂的相特性控制,所述相特性转而主要由脂质组分的性质和比例控制。然而,在本发明情况下,本发明者已确定释放性质,并且特别是在施用之后达到的最大体内浓度(cmax),可通过选择溶剂和溶剂比率来加以有益优化。在一个实施方案中,举例来说,本发明提供本发明的前体制剂,其中组分c)包含乙醇和丙二醇、基本上包含乙醇和丙二醇或由乙醇和丙二醇组成,其中乙醇与pg的比率在1:1与10:1,更优选在1.5:1至8:1,最优选在2:1至5:1之间(例如约3:1)。特定来说,相比于其中相较于pg,存在相等量或较少量的乙醇的制剂,具有乙醇与pg两者(例如各自至少0.5%),其中乙醇的量大于pg的量的制剂可提供较低cmax(即较低“峰值”体内浓度)。对释放性质的所述控制在缓慢释放制剂的情况下具有极大重要性,并且将不被预期会通过选择溶剂比率来提供。关于组分d)的水平,图4a说明tpn从在gdo/pc/etoh(45:45:10)的基质中含有1.53至7.76%之间的tpn(na)的制剂进行的体外释放。在7.76%tpn(na)的装载量下,制剂展现“爆发”特征,即约50%的tpn(na)在约24小时的时期之后释放,而在6.17%以及低于6.17%的水平下,tpn(na)的释放平缓得多。能够仅仅通过适当选择组分来改变爆发概况是本发明的前体制剂的潜在极其适用特征。在一个实施方案中,这些可通过选择溶剂和溶剂比率来影响。在一个实施方案中,特别是当需要的是制剂历经1至3天提供短期释放时,可合乎需要的是以6.5%或更大,尤其以6.7%或更大,或7%或更大的尤其是tpn或tpn(na)的前列环素类似物的水平进行操作。然而,一般来说,前列环素类似物的水平将通常是至多5重量%,优选是至多4重量%(例如0.5至4重量%,诸如约1重量%、约2重量%或约3重量%)。历经1至3天的时期提供有效释放的短期释放储槽可用乙醇作为唯一组分c)以至少11%,优选以至少12%,尤其以至少13%的水平配制。或者,乙醇和亚砜尤其是乙醇和dmso的混合物可以20%或更小,诸如以10至20%,尤其以12至18%的量用作组分c)。在这个实施方案中,乙醇:亚砜的比率在20:80至60:40(w:w)的范围内,尤其在30:70至50:50的范围内。当需要的是提供前列环素类似物的更平缓释放时,例如对于一周时长或每两周或每月持续时间储槽,可合乎需要的是以小于6.5%,诸如以6.2%或更小的d)的水平进行操作,尤其5.5%或更小或5%或更小可为合乎需要的。如上所指示,前列环素类似物的水平将通常是至多5重量%,优选是至多4重量%(例如0.5至4重量%,诸如约1重量%、约2重量%或约3重量%)。因此,用于每周一次或每两周一次施用的前体制剂可优选包含1至7%的前列环素类似物,诸如1至3%,尤其是tpn或tpn(na)。历经大于5天的时期诸如每周或每两周提供有效释放的长期释放储槽可用乙醇作为唯一组分c)在小于11%诸如10%或更小的水平下配制。或者,乙醇和二醇溶剂或多元醇溶剂尤其是乙醇和pg或乙醇和水的混合物可以5至20%,尤其以5至15%的量使用,其中乙醇:pg或乙醇:水的比率在40:60至60:40(w:w)的范围内,约50:50的水平是特别优选的。约2.5%的量的pg和约7.5%的量的乙醇是高度有效的。在一个方面,本文各实施方案可任选含有抗微生物剂或抑微生物剂,其包括抑菌剂和防腐剂。所述试剂包括苯扎氯铵、间甲酚、苯甲醇或其他酚性防腐剂。可使用如本领域中已知的典型浓度。超出提及为组分a)至d)的那些组分的额外组分在确实存在时将优选以0至5重量%(例如0.01%至5%),优选以至多2重量%,并且更优选以至多1重量%的量存在。在一个实施方案中,组分a)和b)(虑及这些组分的在性质方面是固有的任何杂质)构成组合物的脂质组分的至少95%。优选地,前体制剂的总脂质含量的至少99%由组分a)和b)组成。优选地,前体制剂的脂质组分基本上由组分a)和b)组成。施用本发明的前体制剂通常被配制来加以胃肠外施用。这个施用将通常不是血管内方法,但将优选是皮下(s.c.)、腔内或肌肉内(i.m.)方法。重要的是,本发明的前体制剂具有它们不需要在静脉内或通过连续皮下注射来施用的优势。优选地,施用不是静脉内或连续皮下施用。通常,施用将通过注射进行,所述术语在本文中用于指示其中诸如通过针、导管或无针(needle-less/needle-free)注射器来使制剂穿过皮肤的任何方法。优选胃肠外施用通过肌肉内或皮下注射,最优选通过皮下注射来进行。本发明组合物的一重要特征是它可通过肌肉内途径与皮下途径两者以及其他途径来施用,而不具有毒性或显著局部效应,尤其是不导致显著部位疼痛。它也适于腔内施用。相比于(深度)肌肉内注射,皮下注射具有深度较小,以及对受试者的疼痛较小的优势,并且在技术上最适于本发明情况,因为它使易于注射与低局部副作用风险组合。本发明者的一惊人观察结果是通过皮下注射,制剂历经可预测时期提供活性剂的持续释放,并且通常,相较于现有前列环素类似物制剂,本发明制剂使得可获得长久得多的释放持续时间。本发明的优选脂质前体制剂在尤其是在体内暴露于水性流体后提供非层状液晶储槽组合物。如本文所用,术语“非层状”用于指示正液晶相或更优选地反液晶相(诸如反立方相或六方相)或l3相或其任何组合。术语液晶指示所有六方液晶相、所有立方液晶相和/或其所有混合物。除非另外指定,否则如本文所用的六方指示“正”六方或“反”六方(优选是反六方),并且“立方”指示任何立方液晶相。通过参照本文提供的描述和实施例以及参照wo2005/117830,熟练读者在鉴定具有适当相特性的那些组合物方面将不具有困难,但对于相特性来说最有利的组成范围是其中组分a:b的比率如先前章节中所述。约50:50(例如±2)的比率对于大多数制剂是高度优选的,最优选是约50:50。重要的是应了解本发明的前体制剂具有低粘度。因此,这些前体制剂必须不处于任何主体液晶相,因为相比于可通过注射器或类似注射分配器施用的粘度,所有液晶相都具有显著更高的粘度。因此,本发明的前体制剂将处于非液晶状态,诸如溶液、l2或l3相,特别是溶液或l2。如本文整篇所用的l2相优选是“溶胀”l2相,其含有大于5wt%,优选大于7%,并且最优选大于9%的具有降低粘度作用的有机溶剂(组分c)。处于l2相的本发明的前体制剂形成一组优选前体制剂。如本文所用,术语“低粘度混合物”或“低粘度前体制剂”用于指示可易于向受试者施用,并且特定来说易于借助于标准注射器和针装置来施用的混合物。这可例如通过能够通过小规格针从1ml抛弃式注射器分配来指示。优选地,低粘度混合物可通过手动加压通过19规格的针,优选通过小于19规格,更优选通过23规格(或最优选是甚至27规格)针来分配。在一特别优选实施方案中,低粘度混合物应是能够穿过标准无菌过滤膜诸如0.22μm针筒过滤器的混合物。适合粘度的典型范围将是例如在20℃下10至1000mpas,更优选是10至800mpas,并且最优选是200至700mpas。在施用后,本发明的优选的基于脂质的前体制剂经受从低粘度混合物向高粘度(通常具有组织粘附性)储槽组合物的相结构转变。通常,这将是从分子混合物溶胀l2和/或l3相向一种或多种(高粘度)液晶相诸如反六方或立方液晶相或其混合物的转变。其他相变也可在施用之后发生。显然,完全相变不为本发明的运行所必需,但至少所施用混合物的表面层将形成液晶结构。通常,这个转变对于至少所施用制剂的表面区域(与空气、身体表面和/或体液直接接触的那个部分)来说将是快速的。这将最优选是历经数秒或数分钟(例如1秒直至30分钟,优选直至10分钟,更优选是5分钟或更短时间)。组合物的其余部分可通过扩散和/或随着表面区域分散来更缓慢地使相变为液晶相。在不受理论束缚下,据信在暴露于过量水性流体后,本发明的前体制剂丧失一些或全部其中包括的有机溶剂(例如通过扩散),并且从身体环境(例如体内环境)吸收水性流体。对于脂质前体制剂,至少一部分制剂优选产生非层状相结构,特别是液晶相结构。在大多数情况下,这些非层状结构是高度粘性的,并且不易于溶解或分散至体内环境中。结果是在体内产生仅具有有限体液暴露面积的整体“储槽”。此外,因为非层状结构具有大型极性区域、无极性区域和边界区域,所以脂质储槽高度有效使活性剂溶解和稳定,并且保护这些活性剂免遭降解机理。随着由前体制剂形成的储槽组合物历经数天、数周或数月的时期逐渐降解,活性剂逐渐从组合物中释放和/或扩散出来。因为储槽组合物内的环境相对受到保护,所以本发明的前体制剂高度适于具有相对短暂生物半衰期的活性剂。相较于在大致上不存在水下含有有机溶剂的组合物,通过将至少2%(例如至少5%)的极性共溶剂(尤其是至少5%pg、水、nmp或dmso)并入前体制剂中,据信在经注射前体制剂的表面处向非层状(例如液晶)相的相变的速率可得以增强。因此,所得储槽的性能得以改进,并且实现对活性剂的释放的进一步控制。由本发明制剂形成的储槽系统高度有效保护活性剂免遭降解,因此允许释放时期延长。因此,本发明制剂可提供前列环素类似物的体内储槽,其需要仅每1至60天施用一次。典型施用间隔将是例如每1、2、3、7、14、21、28、30或60天,并且可系统地或偶尔地以小量(例如以±3天,或在任何适当情况下以±20%)加以变化。高度优选的施用频率包括每7(±1)天或每14(±2)天,或每30(±3)天。在一个实施方案中,含有相比较来说较低水平的前列环素类似物(例如0.5至2.0%)的制剂可每周一次、每两周一次、或每月一次加以施用,并且具有较高水平的前列环素类似物(例如以重量计,2.5%至4%或更大)的制剂可每周一次或更频繁地诸如每3天一次、每2天一次或每日加以施用。显然,较长稳定释放时期合乎患者舒适性和顺应性的需要,以及如果组合物将不是自我施用型的,那么需要花费健康专业人员较少时间。当组合物将是自我施用型的时,患者顺应性可由每周(例如每7天,任选±1天)或每月(例如每28或30天(任选±7天))施用加以辅助以致不会忘记需要施用。即使是提供不需要连续施用或每日超过一次加以施用的制剂也将在许多情况下在这个领域中极大改进患者康宁。在本发明的一个可适用于所有方面,但特别是可适用于治疗方法和相应用途的实施方案中,施用剂量和频率可逐渐升高以与潜伏疾病(诸如本文指示的那些疾病中的任一者)的进展相对应。因此,1mg/周或5mg/周的剂量的前列环素类似物对于处于早期的受试者可为足够的,并且这些剂量可根据需要以每周、每两周或每月施用的形式提供,例如每4周在10mg/ml下注射1.5ml将给与3.75mg/周的平均剂量,并且对于疾病早期可为足够的。当疾病进展时,剂量可增加至每两周1ml(在10mg/ml下)(5mg/周)、每周0.75ml(7.5mg/周)和1.0ml/周(10mg/周)。后续增加可接着通过较高浓度的制剂来实现,诸如每两周1.0ml30mg/ml制剂(15mg/周),通过使频率和体积增加至例如每周1.0ml(30mg/周)来升高,接着如果必要,那么增加至每周多次施用。在使用连续输注下,已知前列环素类似物的约1至4ng/kg/分钟的初始剂量作为起始输注剂量是典型的,对应于每周约10至40μg/kg,特别是对于依前列醇和它的盐(例如钠盐)来说。所述剂量形成适于本发明制剂的起始剂量,其可接着加以滴定直至实现适合功效/可耐受性平衡。对于曲前列环素(和盐诸如钠盐),适合起始剂量通常是依前列醇的起始剂量的约一半,对应于0.5至2ng/kg/分钟(每周约5至20μg/kg)。再次,可应用向上滴定直至确定适合剂量。本发明的施用方法的一重要方面是尽管本文所述的前体制剂将优选持续至少7天提供前列环素类似物的控制释放,但施用频率可比这个更快速。因此,举例来说,在单次注射本发明的前体制剂之后在7天时前列环素类似物的血浆浓度可下降至在那个施用之后在第1天结束时血浆浓度的不低于10-3,优选不低于10-2,并且最优选不小于10-1。在疾病早期,7天持续时间产品的这种控制释放性能可为足够的,并且在那个方面提供重大优势。然而,相比于每7天,对具有如所描述的释放性质的这种产品的施用可更频繁地加以重复(例如一周两次、每3天、每2天或每日)。这个施用将有意义地发生在第一施用的作用失效之前。然而,多次注射(例如在多个部位处)本文所述的长久持续时间产品提供对前列环素类似物血浆浓度的还要更大调平,并且允许在不需要大注射体积下采用极高剂量。因此,举例来说,每周两次注射1.0ml30mg/ml制剂将在不进行任何大型注射下以及在极其稳定释放概况下提供60mg/周,因为一个释放的“峰值”对应于先前施用的稳定平稳水平。以这个方式,具有名义7天或更久持续时间(例如上述7天释放概况)的本发明的前体制剂可一周两次、每3天、每2天或每日加以使用以在疾病进展要求那样做时提供较高和稳定浓度的前列环素类似物。本发明的储槽前体的另一重大优势是它们是稳定均质相。也就是说,它们可在室内或冰箱温度下储存可观时期(优选是至少6个月,尤其是至少12个月),而不进行相分离。也提供有利储存和轻易施用,这允许通过参照个别受试者的物种、年龄、性别、重量和/或身体状况,借助于注射所选体积来选择前列环素类似物的剂量。因此,本发明提供包括特别是根据受试者重量来选择个体特异性给药量的方法。用于这个剂量选择的手段是选择施用体积。本发明的前体制剂是高度有利的,因为它们在以它们的最终“准备用于施用”形式长期储存时是稳定的。因此,它们可易于被提供来由健康专业人员或由患者或他们的护理者进行施用,这些人无需是经过充分训练的健康专业人员,并且可不具有配制复杂制剂的经验或技能。这在持续时间长久、见效缓慢的疾病诸如糖尿病的情况下是特别重要的。pde5抑制剂在本发明的另一方面,本发明的前体制剂将包含至少一种前列环素或前列环素类似物诸如本文所述的那些,并且可另外包含至少一种pde5抑制剂。pde5抑制剂已通常被施用来即时治疗勃起功能异常(ed),并且已被有利地提供在快速起效制剂中。然而,已在对包括肺动脉高血压(pah)的若干临床重大疾患的长期治疗中指示和/或测试在适合剂量下的pde5抑制剂。因此,可将适合剂量的pde5抑制剂添加至任何本发明制剂中以提供双效治疗。适合pde5抑制剂包括已知抑制剂,诸如阿伐那非(avanafil)、罗地那非(lodenafil)、米罗那非(mirodenafil)、西地那非(sildenafil)、他达那非(tadalafil)、伐地那非(vardenafil)、乌地那非(udenafil)、扎普司特(zaprinast)、淫羊藿苷(icariin)(及其合成衍生物)、本扎那非(benzamidenafil)、达生他非(dasantafil)、其盐、前药和混合物。高度适合的pde5抑制剂包括他达那非(西力士(cialis))和伐地那非(艾力达(levitra))。pde5抑制剂的适于每周一次施用的剂量将通常在1至75mgpde5抑制剂(以游离碱或未保护药物分子计),优选在每周2至50mg(即每次施用),并且最优选在每周5至25mg的范围内。pde5抑制剂的适于每两周一次施用的剂量将通常在2至150mgpde5抑制剂(以游离碱或未保护药物分子计),优选在每两周5至100mg(即每次施用),并且最优选在每两周10至50mg的范围内。pde5抑制剂的适于每月一次施用的剂量将通常在5至300mgpde5抑制剂(以游离碱或未保护药物分子计),优选在每月10至200mg(即每次施用),并且最优选在每月20至100mg的范围内。或者,本发明的储槽前体可与包含至少一种pde5抑制剂的等效制剂相伴施用(即一个施用是含有前列环素类似物的前体制剂,并且另一施用是含有pde5抑制剂的前体制剂)。所述相伴施用可为同时的,或以任一顺序依序的,但将通常在同一天,并且采用具有类似持续时间的储槽前体制剂(例如两者均将是每月制剂或两者均是每周制剂)。优选地,相伴施用将是施用包含类似控制释放基质(例如两者均包含脂质或两者均包含聚合物,诸如任何本文指示的优选系统)的前体制剂,并且最优选地,将是施用均是脂质制剂,并且包含相同或大致上相同的脂质组分(任选以及相同或大致上相同的溶剂组分)的前体制剂。最优选组分将包括如本文所述的dag(例如gdo)和磷脂(例如pc)。装置在另一方面,本发明提供一种用经计量剂量的本发明的前体制剂预装载的抛弃式施用装置(其也将包括装置组件)。这种装置将通常含有准备用于施用的单次剂量,并且将通常加以无菌包装以使组合物储存在装置内直至施用。适合装置包括药筒、安瓿,并且特别是注射器和注射器筒体,具有整体针或具有适合于采用适合抛弃式针的标准(例如鲁尔(luer))接头。类似适当的装置包括无针注射器、多次使用或单次使用自动注射器与预填充注射器组合、药筒任选与多次使用笔装置组合、或小瓶。显然,所述预填充注射器和药筒可用于任何适当注射装置,诸如多次使用或单次使用注射器或无针注射装置。本发明装置可优选含有递送在2至50mg/ml,优选在5至40mg/ml,最优选在7至35或10至30mg/ml的范围内的剂量的本发明的前体制剂。剂量体积将通常是至多2ml(例如0.1至2ml),例如0.25至1.5ml或0.5至1ml。每周或更经常施用的制剂在体积方面可优选是至多1.2ml或1.0ml,其中每两周或每月施用的制剂在体积方面可优选是至多2ml或1.5ml。在一个可适用于本发明的所有方面的实施方案中,本发明装置可含有1至200mg例如2至150mg(例如5至120mg)的单次剂量的前列环素类似物。本发明装置可含有在约0.005至2.5mg/kg/周下,优选在0.01至1mg/kg/周,尤其在0.015至0.7mg/kg/周的水平下的前列环素类似物。用于50kg、70kg或80kg受试者以及所有其他受试者或受试者重量范围的剂量可相应地加以计算。举例来说,前列环素类似物的适于70kg受试者的剂量将在0.35至175mg/周,优选在0.7至70mg/周,尤其在1至50mg/周的范围内。本发明装置可含有至多2ml,优选是至多1ml,尤其是至多0.5ml的总施用体积。本发明的预填充装置也可适合地包括在施用药盒中,所述药盒也形成本发明的另一方面。在另一方面,因此,本发明提供一种用于施用至少一种前列环素类似物的药盒,所述药盒含有经计量剂量的本发明制剂以及任选施用装置或其组件。优选地,剂量将被容纳在将适于肌肉内或优选皮下施用的装置或组件内。药盒可包括额外施用组件诸如针、拭子等,并且将任选和优选含有施用说明书。所述说明书将通常涉及通过如本文所述的途径进行的施用和/或对本文在以上指示的疾病的治疗。药盒本发明提供包含如本文所述的前体制剂的一种如本文指示的预填充施用装置和一种如本文指示的药盒。适合药盒可包括单次使用或多次使用注射装置诸如自动注射器,或可包括用于所述装置中的药筒或组件。本发明药盒将另外(任选但优选)包括任何以下组件:i)注射装置,诸如注射器或自动注射器ii)剂量计量装置(例如用于计量或设置施用体积的刻度装置)iii)用于基于诸如受试者重量和/或剂量频率的参数来计算和/或设置剂量体积的表、图、电话应用程序或电子计算器。在所述计算中,可明确地或隐含地考虑诸如疾病进展和/或前列环素类似物浓度的因素。iv)关于根据诸如受试者重量和/或剂量频率、疾病进展(例如平均肺动脉压)和/或前列环素类似物浓度的因素进行给药和/或逐步升高给药的说明书。优选特征和组合与本文指示的特征和优选特征相组合,本发明的前体制剂可独立地或组合地具有一种或多种以下优选特征:本文指示的所有比例都可任选变化指定量的多达10%,任选和优选多达5%;组分a)包含gdo、基本上由gdo组成、或优选由gdo组成;组分b)包含以下、基本上由以下组成、或优选由以下组成:大豆pc和/或“高纯度pc”诸如dopc;组分c)包含以下、基本上由以下组成或由以下组成:1、2、3或4碳醇,优选是异丙醇,或更优选是乙醇;组分c)包含极性溶剂诸如水、nmp、dmso、丙二醇或其混合物;前体制剂具有如本文指示的低粘度。前体制剂在体内施用后形成如本文指示的液晶相。前体制剂在体内施用之后产生储槽,所述储槽历经至少3天,优选至少5天,更优选至少7天的时期释放至少一种前列环素类似物。前体制剂在向受试者进行体内施用之后产生储槽,所述储槽释放至少一种前列环素类似物以使在施用之后在第七天结束时所述受试者中前列环素类似物的血浆浓度是在施用之后在第一天结束时(即在施用之后24小时时)所述受试者中前列环素类似物的血浆浓度的不小于10-4倍或10-3倍,优选不小于10-2倍,更优选不小于10-1倍。与本文指示的特征和优选特征相组合,本发明的治疗方法可独立地或组合地具有一种或多种以下优选特征:方法包括施用至少一种具有一种或多种如上指示的优选特征的制剂;方法包括通过肌肉内注射、皮下注射或优选通过深度皮下注射来施用至少一种如本文指示的制剂;方法包括借助于如本文指示的预填充施用装置进行施用;方法包括通过不大于20规格,优选小于20规格,并且最优选是23规格或更小的针来施用;方法包括每3至10天,优选每5至8天进行单次施用。方法包括随着疾病(本文指示的那些)的进展逐步升高剂量和频率,以使频率从至多每周逐步升高至至少每3天施用,并且从至多10或20mg/周逐步升高至至少30或40mg/周。与本文指示的特征和优选特征相组合,本文指示的前体制剂制造药剂的用途可独立地或组合地具有一种或多种以下优选特征:用途包括至少一种具有一种或多种如上指示的优选特征的制剂的用途;用途包括制造用于通过肌肉内注射、皮下注射或优选通过深度皮下注射来施用至少一种如本文指示的制剂的药剂;用途包括制造用于借助于如本文指示的预填充施用装置进行施用的药剂;用途包括制造用于通过不大于20规格,优选小于20规格,并且最优选是23规格或更小的针来施用的药剂;用途包括制造用于每日或每2、3、7、14、21、28、30或60天一次施用的药剂,并且可系统地或偶尔地以小量(例如以±3天,或在任何适当情况下以±20%)加以变化。与本文指示的特征和优选特征相组合,本发明的预填充装置可独立地或组合地具有一种或多种以下优选特征:它们含有如本文指示的优选制剂;它们包括小于20规格,优选不大于23规格的针;它们含有单次剂量的在2.5至50mg/ml下的前列环素类似物(基于游离酸),诸如5至50mg/ml(基于游离酸)。它们含有具有本发明的组成的呈准备用于注射形式的均质混合物。它们含有组分a)至c)的用于与前列环素类似物组合,借此以形成本发明的前体制剂的制剂。它们含有至多5ml,优选至多3ml,例如至多2ml,更优选至多1.5ml的总施用体积。与本文指示的特征和优选特征相组合,本发明的药盒可独立地或组合地具有一种或多种以下优选特征:它们含有如本文指示的优选制剂;它们含有如本文指示的预填充装置;它们含有小于20规格,优选不大于23规格的针;它们含有1至100mg的单次剂量的前列环素类似物(如本文所述),优选是2至75mg,更优选是3.5至60mg。它们含有“双区室药盒”,其包括至少两个分别含有本发明的脂质制剂和前列环素类似物的容器。它们含有至多5ml,优选至多3ml,例如至多2ml,更优选至多1.5ml的总施用体积。它们含有关于通过如本文指示的途径和/或在如本文指示的频率下施用的说明书;它们含有关于在如本文所述的治疗方法中使用的施用说明书。如本文所用,关于数值或数值范围的术语“大约”、“约”、“大致上”或“近似”将通常指示指定的数值或范围是优选的,但这种数值可在某一程度上变化而不实质上影响相关物质、组合物或类似产品的性质。熟练工作者将通常能够易于确定所述数值可加以变化而不损害本发明的关键优势所处的程度。作为一般准则,所述数值或所述范围的端值可变化±10%,优选±5%,并且更优选±1%。相应含义可归于“基本上由某些组分组成”的组合物,其除指定的那些组分之外也可包括多达10%,优选多达5%,并且最优选多达1%的其他组分。当化学基团、链或其他部分在本文中被描述为任选被取代时,所述取代可不存在,或所述部分中的一个或多个原子(通常是一个或多个氢和/或碳)可被诸如以下的基团取代:卤素(例如f、cl、br、i)基团,基于氧的部分诸如醚、醇、酯、羧酸或环氧化物,基于氮的基团诸如胺基团、酰胺基团、腈基团或硝基,或基于硫的基团诸如硫醇、二硫醚、硫酯等。当上下文允许时,可进行多达约10个所述取代,但通常用独立选择的取代基团进行的3个取代或少许取代诸如1、2或3个取代将是典型的。本发明现将通过参照以下非限制性实施例和附图来进一步说明。实施例材料来自sanofi的曲前列环素钠盐tpn(na);来自lipoid的大豆磷脂酰胆碱spclipoids100;来自croda的甘油二油酸酯gdocithrolgdohp-so-(lk);来自nof的二油酰基磷脂酰胆碱dopc;来自solveco的乙醇etoh(99.7%ph.eur);来自fischer的丙二醇pg(ph.eur);来自sigma-aldrich的n-甲基吡咯烷酮nmp和二甲亚砜dmso按原样加以使用。所有其他化学物质都具有分析级纯度。制备前体制剂通过将适量的spc、gdo和溶剂称重至灭菌玻璃小瓶中来制备脂质储备混合物。接着将密封小瓶在室温(rt)下放置在辊式混合器上直至完全混合成澄清均质液体溶液(<24小时)。在新玻璃小瓶中将tpn(na)粉末添加至相应脂质安慰剂制剂中。接着将小瓶密封,并且在室温下放置在辊式混合器上直至完全混合成澄清均质液体溶液(<24小时)。将制备的制剂在室温下在黑暗中储存直至进一步实验。对于探索性稳定性评估,将制剂分装至灭菌2r玻璃小瓶中(每个小瓶1g制剂)。将小瓶密封,并且放置在受控环境储存橱柜中。在预定取样点,从各储存橱柜取出两个小瓶的制剂,在室温下放置1小时,并且使用梯度hplc以uv检测来分析含量和纯度。实施例1:评估tpn(na)溶解性和体外释放通过以下方式来评估溶解性:将tpn(na)添加至相应脂质储备混合物中,随后在室温(rt)下在辊式混合器上混合直至完全混合成澄清均质液体溶液。在制备期间,对样品进行目视检查。结果显示tpn(na)在多种前体制剂中具有良好溶解性,并且至少7wt%(约78mgtpn(0)/ml)的药物装载量是可行的。如表1中所示,视共溶剂类型、浓度和组成而定,制剂的测量粘度在185-628mpas之间的范围内。表1.含有7wt%tpn(na)的各种前体制剂的粘度。使用基于uv/vis光谱法的简明定量测定对tpn(na)进行体外释放测试。在测试中,通过将0.03-0.10g(靶标值0.1g)相应前体制剂注射至保持在20r玻璃注射小瓶中的10mlpbs(ph7.4)中来制备储槽。添加至各小瓶中的制剂的精确量通过称重来确定。将小瓶用橡胶塞和铝压接盖密封,并且在振荡下放置在保持在37℃下的孵育器中。在预定时间点对释放介质取样,稀释并转移至石英比色皿中,并且在perkinelmerlambda25双光束uv-vis分光光度计上在273nm下分析。来自体外释放测量的结果显示于图1a中。根据结果,明显的是溶剂量与组成两者均影响曲前列环素的初始体外释放。相比于具有dmso的制剂,包含pg作为共溶剂的制剂具有较慢初始释放(24小时)。此外,当比较具有etoh作为唯一溶剂的制剂时,在较高溶剂含量下,初始释放较快。图1b进一步指示体外释放是两阶段的,在其中溶剂和药物相伴释放的初始阶段之后,释放与时间的平方根成线性关系,如通过来自整体储槽基质的扩散控制释放机理所预期。实施例2:在大鼠中施用具有tpn(na)的前体制剂:制剂和体重变化这个试探性研究的主要目标在于评估在将具有tpn(na)的前体制剂单次皮下注射至大鼠中之后tpn的局部和全身可耐受性(制剂组成在表2中给出)。研究被设计为剂量逐步升高研究,其中施用剂量是3、9和27mg/kgtpn(表3)。表2.用于试探性大鼠研究中的制剂组成。表3.用于试探性研究中的治疗组和tpn剂量。图2显示在研究期间的平均相对体重变化。如下在试探性研究中监测制剂b1和b2的注射部位红斑/水肿:评分分类0无红斑/水肿1轻微红斑/水肿(依稀可辨)2中度明确确定红斑/水肿3重度红斑至轻微焦痂形成/重度水肿在施用制剂b1和b2之后的红斑和水肿形成程度指示在下表4中:表4.在试探性研究期间在注射部位处的红斑和水肿的概述。如下在试探性研究中监测制剂b1和b2的血管生成/出血:血管生成的水平通过0至3的范围来确定:0代表无血管生成。1代表轻度血管生成。有限的血管生长。2代表中等血管生成。扩大的血管生长。3代表重度血管生成。广泛的血管生长。出血的水平通过0至3的范围来确定:0代表无出血。1代表轻度出血。一个或多个扩散红色区域。2代表中等出血。至少一个明确确定的红色区域。3代表重度出血。若干明确确定的红色区域。在施用制剂b1和b2之后的血管生成和出血程度指示在下表5中。表5.在试探性研究中在尸检时获得的注射部位研究结果的概述。实施例3:对完全水合前体制剂的纳米结构的影响随不同量的tpn(na)而变化制备以下制剂l至aa,其组成和测量粘度分别在表6和图3中给出。表6.用于纳米结构评估的制剂代码和组成。使用小角度x射线衍射评估来自表6的完全水合制剂的纳米结构。简要来说,将约100mg制剂注射至5mlpbs缓冲液中,并且留置以在静止小瓶中在环境室温下平衡8天,随后进行saxd测量。使用同步加速器saxd测量来研究随tpn(na)浓度而变化的完全水合制剂的纳米结构,所述测量使用含有总计981x1043像素的1mpilatus2d检测器(dectris)在maxiv实验室(在1.5gev下运行的maxii电子加速器,lunduniversity,sweden)处在i911-4光束线下进行。将样品在样品与检测器距离1919.5mm下安放在定制钢样品夹持器中薄聚酰亚胺膜之间。采用x射线波长和在样品处光束截面0.25×0.25mm(半高宽)记录衍射图。使用计算机控制的laudare420g恒温器(lauda-brinkmann,lp)实现温度控制。相继在25、37和42℃下,在各温度下采用60s暴露时间,以及在温度阶跃之间等待10分钟来进行实验。所得ccd图像使用由esrf(欧洲同步加速器辐射研究室(europeansynchrotronradiationfacility),france)提供的fit2d软件来积分和分析。使用山嵛酸银校正的样品与检测器距离和检测器位置。图5和图6显示随tpn浓度和温度而变化的完全水合脂质/etoh(90/10wt%)和脂质/etoh/pg(85/7.5/7.5)制剂的纳米结构的所得saxd结果。图5中的数据显示在37-42℃的温度区域中,直至3.1wt%的tpn,完全水合的基于10%etoh的制剂形成反六方(h2)相和反胶束立方(fd3m)相的混合物。在4.65和6.2wt%的tpn下,形成单一h2,其在甚至更高浓度的tpn下开始转变成无序胶束溶液(l2)。此外,随着tpn浓度增加,fd3m相的晶格参数保持不变,而h2相的晶格参数开始增加。从4.65wt%的tpn(na)开始的晶格参数增加可能与在胶凝实验中观察到的储槽机械软度增加相关。总之,所观察到的随着tnp浓度增加从fd3m和h2的混合物向单一h2以及进一步向h2相和l2相的混合物的转变与所得体外释放结果相关联,其中在6.2和7.75wt%的tpn下发现所释放tpn急剧增加(图4)。作为比较,图6显示对在7.5/7.5wt%的etoh/pg混合物下制备的完全水合制剂获得的saxd结果。在此处,在37-42℃的温度区域中,仅直至1.55wt%的tpn,形成fd3m相和h2相的明确断定混合物。在2.33wt%tpn与6.2wt%tpn之间增加tpn浓度,观察到h2相与l2相的混合物。此外,在7.75wt%的tpn下,形成层状(lα)相和l2相的混合物。此外,在3.10wt%的tpn(na)下,六方相的晶格参数已开始增加。基于这些结果,可推断使用7.5/7.5etoh/pg制备的完全水合制剂在转变成从持续释放的观点来看较不有利的更溶胀液晶相(尤其是lα)之前可容纳较少tpn。测量制剂n、p、q、r和s的体外释放概况,并且显示于图4a-图4b中(累积释放%)。实施例4:含有tpn(na)的前体制剂的物理和化学稳定性在以下条件下通过hplc来研究制剂bb、cc和dd的储存稳定性(表7和表8):≤-25℃(冷冻条件);25℃/60%rh;和40℃/75%rh。来自探索性稳定性测试的结果指示使用10%etoh作为溶剂的前体制剂以及具有etoh/pg的混合物的前体制剂中的tpn均具有良好物理以及化学稳定性。当在25℃/60%rh和40℃/75%rh下储存时,tpn显示持续至少多达3个月的良好稳定性(表9)。表7.用于物理和化学稳定性评估的制剂代码和组成。表8.用于物理和化学稳定性评估的制剂代码和粘度。表9.在来自探索性稳定性研究的制剂的情况下在不同储存条件下随时间变化的tpn测定结果(hplc)。实施例5:在大鼠中施用具有tpn(na)的前体制剂使用不同溶剂组成制备制剂ee-hh(表10),并且向一组24只大鼠施用并使用较早实施例的评分系统监测红斑、水肿、血管生成和出血。表10.用于大鼠pk研究中的制剂组成和tpn剂量。表11.在施用制剂ee-hh之后的红斑程度。表12.在施用制剂ee-hh之后的水肿程度。表13.在施用制剂ee-hh之后在尸检时的注射部位。实施例6:具有tpn(na)的前体制剂的大鼠pk数据。历经14天收集实施例7的向大鼠施用的制剂ee至hh的药物动力学数据。数据以图解方式说明于图7中,并且相应体外释放概况说明于图8中。表14.制剂ee-hh在大鼠中的pk参数。数据以图解方式说明于图10中,并且释放百分比概况说明于图11中。实施例7:在狗中皮下注射具有tpn(na)的前体制剂这个研究的目标在于在最大耐受剂量毒性研究中评估在将具有tpn(na)的前体制剂(制剂代码jj,组成tpn(na)/gdo/spc/etoh/pg3.38/43.31/43.31/7.50/2.50wt%)皮下注射至比格犬中之后的tpn暴露。用于狗研究中的tpn剂量(以tpn酸形式形式计算)在表15中给出。所得剂量依赖性pk概况和暴露(auc0-168h)值分别呈现于图9和图10中。表15.用于狗pk研究中的tpn剂量水平和体积。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1