综合应用第三代超长测序读段和第二代链接式读段从头组装基因组的方法与流程
本发明涉及基因组测序数据从头组装的方法,特别是第三代测序数据和第二代测序数据的混合组装方法。第三代测序数据主要是由pacbio、nanopore或其他测序技术产生的超长读段,第二代测序数据主要是10xgenomics测序产生的链接式读段。结合高效率的组装软件——dbg2olc,极大的降低了测序成本和计算成本(尤其是第三代测序技术的应用成本)。为应用第三代测序技术进行大规模、高质量的基因组从头组装提供了一种高效、可靠的方法
背景技术:
随着测序技术的发展,基因组从头组装产生的基因组序列信息越来越详细、准确。近年来发展出来的10xgenomics测序技术更是能够以较低成本的从头组装出同源染色体。传统的第二代测序技术产生的测序短读段(150-300bp)难以解决基因组中大量重复序列的组装,重复序列使得组装产生大量的短片段。10x测序技术在illumina测序技术的基础上,给待测基因片段加入标签(barcoding),该标签标记了测序读段的来源(链接式读段),极大的降低了基因组装的复杂度,并且避免了不同来源的测序读段错配的可能。10x测序技术解决了传统第二代测序技术的重复序列带来的问题,能够组装出更长的基因组草图。
第三代测序技术克服了传统第二代测序技术的缺陷,可以产生超长的测序读段。太平洋生物科学公司的pacbio测序平台和英国牛津纳米孔测序公司的nanopore测序平台是目前应用最为广泛的第三代测序技术。它产生的超长读段也能解决基因组中重复序列的拼接问题。其中nanopore测序技术产生的读段长度达到几百kbp,甚至能达到mbp级别。
但是第三代测序技术的高错误率带来的组装问题成为其大范围推广的障碍。第三代测序数据的错误位点是随机发生,这种错误可以通过提高测序覆盖度(coverage)纠错,但是覆盖度的增加导致测序数据的增加,从而增加了测序测序成本和计算成本。尽管pacbio和nanopore测序技术已被成功应用于基因组的从头测序,但是高昂的测序成本和计算成本阻碍了第三代测序技术的大规模应用。
综合应用第二代测序数据和第三代测序数据的混合组装策略在一定程度上解决了三代数据高错误率和二代数据读段短的缺陷。二代测序数据和三代测序数据互相取长补短,在保证组装质量的基础上,提高了组装效率和准确程度,降低了测序成本和计算成本。针对混合组装策略开发的dbg2olc软件就是一款高效率的混合组装软件。dbg2olc软件是发明人参与共同开发的高效率混合组装软件(发明名称:用于组装基因组序的方法、系统及装置,申请号201510084489x,已进入实质审查阶段,于2016年10月5日公布)。本发明使用dbg2olc软件组装第三代测序技术产生的超长读段和10xgenomics测序产生的链接式读段,综合二者之长,极大的降低了测序成本和计算成本。
技术实现要素:
本发明的目的在于:
提供一种综合应用第三代测序数据的超长读段和第二代10xgenomics的链接式读段高效低成本从头组装基因组的方法。由于该方法较低的测序和计算成本,可以大规模用于种群或群落多物种基因组从头(denovo)测序和组装,为第三代测序技术的推广提供了技术支持。
本发明采用的技术方案为:
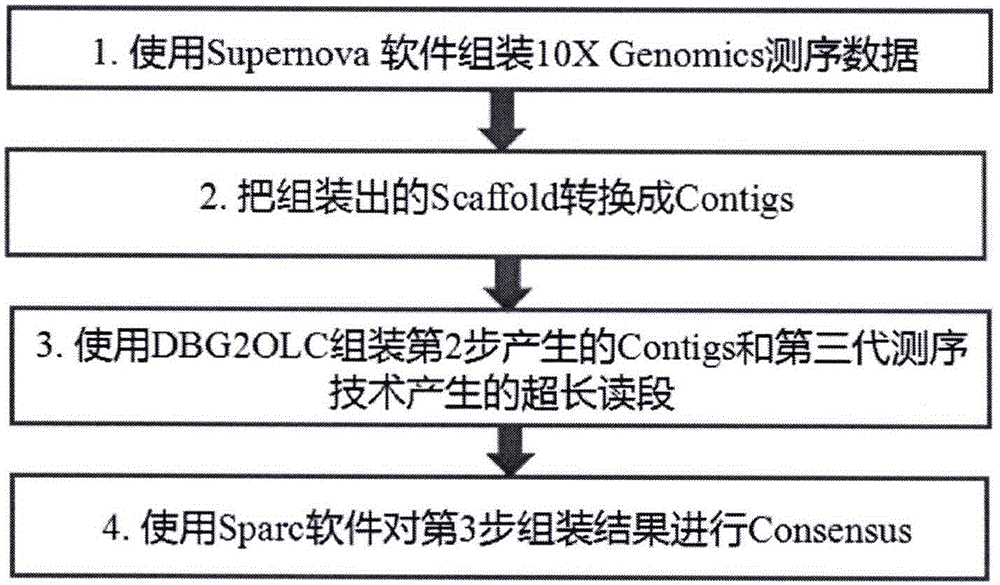
本发明综合应用第三代测序数据的超长读段和第二代10xgenomics的链接式读段高效低成本从头组装基因组,主要技术过程分为4步(见图1):
(1)应用supernova软件组装10xgenomics链接式读段。10xgenomics测序平台采用条形码标记不同的dna待测片段,测序产生的链接式读段特点是同一来源的读段可以通过条形码再次拼接在一起,避免了不同dna片段之间发生错配,极大的降低了计算的复杂程度。该软件直接组装出scaffold。
(2)把supernova组装出的scaffold转换成contigs。把上一步组装出的scaffold重新转换为contigs,用于下一步dbg2olc组装(dbg2olc组装需要使用contigs数据,不能输入scaffold数据)
(3)使用dbg2olc组装第(2)产生的contigs和第三代测序技术产生的超长读段。dbg2olc是一款高效率混合组装第二代和第三代测序数据的软件。该软件需要第二代测序数据组装出的contigs和第三代测序的原始数据。通过第二代测序数据对第三代测序数据进行纠错。在这个过程中,10xgenomics组装出的长contigs使得第三代测序数据计算重叠区的过程更为高效可靠,从而极大的改进了组装效果和计算效率。
(4)使用sparc软件对第(3)步组装结果进行consensus。
本发明的效果在于:
提供一种综合应用第三代测序数据的超长读段和第二代10xgenomics的链接式读段高效低成本从头组装基因组的方法。该方法充分利用了第二代10xgenomics测序链接式读段的优势以及第三代超长读段,通过高效率的混合组装软件dbg2olc组装出高质量的基因组序列。该方法较低的测序成本和计算成本以及高质量的组装结果为第三代测序数据的应用奠定了基础。
附图说明
图1是本发明计算过程的流程图,主要分为四个步骤。涉及到的软件主要是supernova、dbg2olc和sparc三个软件,supernova是专门用于组装10xgenomics链接式读段的软件。dbg2olc是高效组装二代和三代数据的软件,sparc是高效率进行长读段比对的软件,以此为基础对组装结果进行consensus。
图2是本发明所用数据的测序成本。横坐标表示测序覆盖度,纵坐标表示测序成本(单位美元)。图中细线表示用35xnanopore测序数据的测序成本,粗线表示56x链接式读段+7xnanopore测序读段的测序成本(本发明的使用的测序数据)。从图中可以看出本发明测序方案的测序成本远低于35xnanopore测序数据的测序成本。
图3是本发明所使用的方法与35xnanopore测序数据组装结果的比较。把组装后的contigs按照长度从大到小排序,从第一条contigs逐条累加。图3中横坐标表示contigs的编号,纵坐标表示累加序列的长度。图3中粗线表示本发明的方法即56x链接式读段+7xnanopore测序读段组装结果的累加曲线,细线表示35xnanopore测序数据组装结果的累加曲线。从图3中可以看出当累加至第215条contigs序列时,两种方法contigs累加长度相等。图中标记了n25、n50、n75的contigs长度和序号。
具体实施方式
我们采用人类基因验证本发明方法的效果。采用56x的第二代链接式读段测序数据通过supernova组装出scaffold,然后将其转换为contigs,另外选用7x第三代测序超长读段。使用dbg2olc混合组装contigs和7x的超长读段。另外我们采用第二代链接式读段的组装结果、30x和35x的第三代测序读段组装结果用于比较本发明的组装效果和测序成本(结果见表1)。
表1.10xgenomics链接式读段组装结果、nanopore测序数据组装结果和混合组装结果的比较
**本发明使用的方法
表1中总长度是组装出的基因组序列总体长度,人的总长度为3,000,000,000bp,这个值越大说明组装出的基因越完整。序列数量表示组装出的contigs或者scaffold的数量。我们的方法组装出的基因组长度仅次于nanopore(35x)的结果,而且组装出的contigsnumber最少,因此我们的结果做出的序列长度的平均数和中位数最大。说明我们组装出的contigs都比较长,没有特别短的序列。
表1中最长读段表示组装出的最长contigs的长度,这是衡量组装效果的重要指标。表1中10x的scaffold的值最大,但是scaffold中存在间隔(gap)(即两条contigs之间的缺失片段,该片段未测序具体序列,但是已知这段序列的长度,一般用已知长度的“n”补全这一段)。其次是nanopore(35x)的组装结果,这是三代长读段测序的优势,它能组装出较长的contigs,而且没有间隔。我们的方法做出的最长contigs的值比nanopore(35x)的结果短了约30%,但是我们仅用了7x的nanopore数据(用了nanopore(35x)数据的1/5),并且高于nanopore(30x)的结果。
表1中n50,n80,n90,三个值都反映了组装出的contigs长度的分布情况。把所有contigs按照长度从大到小排序,然后从最长的contigs开始累加,当累加长度超过总长度的50%、80%、90%时,最后累加上的contigs的长度即是n50,n80,n90的值。表1中,三个值最大的都是10x的组装结果,与最长contigs长度的问题相同,他们的序列中存在间隔。我们的方法做出的三个值都高于nanopore(30x)的结果。虽然我们的结果中n50的值略小于nanopore(35x)的结果,但是n80,n90的值都高于nanopore(35x)的结果。
- 还没有人留言评论。精彩留言会获得点赞!