高分辨率等位基因鉴定的制作方法
相关专利申请
本申请要求于2013年10月15日提交的临时专利申请no.61/891,193的优先权,该临时专利申请以引用的方式全文并入。
背景技术:
虽然大多数的人基因组由基本上整个人类种群所共有的保守序列构成,但是基因组的小的但是重要的一部分是高度可变的。这些序列差异并未均匀散布在整个基因组中。而是,某些基因组区域(“基因座”)比其他区域含有更多的序列变异(“多态性”)。特定基因座处的具体核苷酸序列(即该基因座处存在的等位基因)的种类可具有重要的生物学意义。例如,个体在特定基因座所携带的等位基因可影响个体是否对疾病具有易感性或者是否治疗剂有可能有效。此外,有关高度多态性基因座处的等位基因的种类的知识可用于追踪生物样品的种族起源和/或地理起源,这对人类学家来说可能是非常有价值的并且这在法医学上可用于将个体与生物样品联系起来。考虑到下一代测序技术的可用性不断增加,将下一代测序数据用于等位基因鉴定的前景颇具吸引力。遗憾的是,利用测序数据来精确且有效地鉴定高度多态性基因座处存在的等位基因是有挑战性的,尤其是在测序数据是使用高通量的基因组范围的测序方法来生成时。
对高度精确的等位基因预测方法存在需求的一套高度多态性基因座是那些编码人白细胞抗原(hla)蛋白的基因座。hla蛋白将抗原肽递呈给淋巴细胞以便介导关键的免疫学事件,包括自体抗原耐受和对病原体或肿瘤的免疫应答。i类hla由所有有核细胞遍在表达,将胞质抗原递呈给细胞毒性t细胞。ii类hla主要由免疫细胞表达,将胞外抗原递呈给辅助t细胞。
人类具有六种主要的hla蛋白,即三种i类蛋白(hla-a、hla-b和hla-c)和三种ii类蛋白(hla-dq、hla-dr和hla-dp)。每种i类蛋白由单个hla基因座(例如,hla-a基因座、hla-b基因座和hla-c基因座)编码。另一方面,ii类蛋白为由α链和β链构成的异型二聚体,这两条链每一者由其自己的hla基因座(例如,hla-dqa1基因座、hla-dqb1基因座、hla-dra基因座、hla-drb1基因座、hla-drb3基因座、hla-drb4基因座、hla-drb5基因座、hla-dpa1基因座和hla-dpb1基因座)编码。在人类中,主要hla基因座(i类和ii类二者)每一者均存在于染色体6上。由于人类是二倍体生物,所以人类携带两个拷贝的染色体6,并因而携带两个拷贝的每种hla基因座。
hla基因座是高度多态性的。hla基因座中的多态性通常导致hla蛋白的氨基酸序列中存在差异。这种hla多样性使得在群体内能将宽泛范围的不同抗原递呈给免疫细胞。然而,hla序列中的这些变异也会导致个体之间器官和组织的组织不相容性,极大地增加了外科移植手术的复杂性。如果由移植的器官或组织表达的hla蛋白被移植接收者的免疫系统识别为外来物的话,则可能的结果是器官排斥。类似地,包括转移将移植接收者中的细胞所表达的hla蛋白识别为外来物的免疫细胞的移植可导致移植物抗宿主病。如果存在于有前景的供应者和接收者的hla基因座处的等位基因编码匹配的hla蛋白的话,则会在最大可能性程度上降低移植物抗宿主病以及器官或组织排斥的风险。为了确定是否存在匹配,有必要确定供应者和接收者中的hla基因座处存在哪种hla等位基因,这是称为hla分型的方法。个体在hla基因座处的hla类型由存在于该个体两个拷贝的hla基因座处的两个hla等位基因(或如果是纯合子的话,两个拷贝的单种hla等位基因)构成。
也越来越认识到hla类型在许多疾病中发挥重要作用。例如,某些hla类型与自身免疫障碍(包括狼疮、炎性肠病、多发性硬化症、关节炎和i型糖尿病)之间存在着强的关联性(例如,graham等人,eur.hum.genet.(《欧洲人类遗传杂志》)15:823-830(2007);fu等人,j.autoimmun.(《自身免疫杂志》)37:104-112(2011);cassinotti等人,am.j.gastroenterol(《美国胃肠道杂志》)104:195-217(2009);luckey等人,j.autoimmun.(《自身免疫杂志》)37:122-128(2011);lemire,m.,bmcproc.(《bmc汇刊》)7:s33(2009);noble等人,curr.diab.rep.(《现代糖尿病报道》)11:533-542(2011),特此将这些参考文献每一者以引用的方式全文并入)。举个例子,ii类hladqa1*02:01(dq2)和drb1*03:01(dr3)常常存在于全身性红斑狼疮患者中并且与疾病易感性显著相关联(graham等人,eur.hum.genet.(《欧洲人类遗传学杂志》)15:823-830(2007))。其他ii类hla蛋白的存在也与乳腺癌和宫颈癌的耐受性或易感性相关(例如,chaudhuri等人,proc.nuc.acad.sci.usa(《美国科学院院刊》)97:11451-11454(2000);garcia-corona等人,arch.dermatol《皮肤病学纪要》.140:1227-1231(2004),特此将这些文献的每一者全文以引用的方式并入)。
hla分子的发病机理和治疗适应症凸显了对精确且有效的hla分型方法的需求。过去,hla类型通过区分“两位数(two-digit)”抗原组团以低分辨率解析,“两位数”抗原组近似于肽结合中的血清学特异性。然而,对于许多应用,两位数hla分型是不够的。例如,具有相同两位数类型的两种hla蛋白质之间的单氨基酸差异可导致改变的t细胞识别特异性和组织排斥(例如,archbold等人,trendsimmunol.(《免疫性趋势》)29:220-226(2008);tynan等人,nat.immunol.(《自然免疫学》)6:1114-1122(2005);fleischhauer等人,neng.j.med.(《新英格兰医学杂志》)323:1818-1822(1990),特此将这些参考文献的每一者以引用的方式全文并入)。因此,在氨基酸序列水平上的高分辨率hla分型(也称为“四位数”分型)可能是关键的。例如,以高分辨率解析hla类型可实质上改善不相关脐带血移植中以及癌症疫苗接种试验中的临床结果(nagorson等人,cancerimmunol.immunother.(《癌免疫学和免疫疗法》)57:1903-1910(2008);liao等人,bonemarrowtransplant.(《骨髓移植》)40:201-208(2007),特此将这些参考文献的每一者以引用的方式全文并入)。
hla基因座的高度多态性性质使得精确、高分辨率分型存在相当大的挑战,尤其是以高通量进行分型时。在人种群中,主要的i类和ii类hla基因座处存在超过7527种四位数hla等位基因。能够以四位数分辨率解析hla类型的现有hla分型方法(如通过测序特异性引物(ssp)进行的群组特异性pcr(groupspecificpcrbysequencingspecificpriming))和基于序列的分型(sequence-basedtyping,sbt))具有低的通量。其他提出的分型策略通过pcr扩增特异性地靶向hla基因座,然后进行深度测序。这种方法要求长的读段和高的覆盖度(深度)以便产生对四位数hla等位基因的精确赋值。由于费用和效率的考虑,基因组范围的测序(如转录组测序或全外显子/基因组测序)通常产生短很多的读段(<100个碱基)和低很多的覆盖度。这些读段长度和覆盖度局限性降低了试图将基因组范围的测序方法用于hla分型的当前方法的精确度。具体而言,据报道,利用短读段测序的当前方法的四位数hla类型鉴定的精确度介于32%到84%之间(例如,boegel等人,genomemed.(《基因组医学》)4:102(2013);kim和pourmandplosone((《公共科学图书馆·综合》),8:e67885(2013))。
鉴于以上所述,存在对新方法的需求,这种新方法利用多样的测序数据(包括具有短的读段长度和低的序列覆盖度的数据)精确且有效地鉴定基因座处存在的等位基因。
技术实现要素:
在一些方面,本文提供用于精确地确定基因座处存在的等位基因(例如,确定hla基因座处的hla类型)的方法(包括计算机实现的方法)、计算机程序和计算机系统。本文还提供用于移植器官、组织或细胞的方法、用于防止移植排斥的方法和/或用于防止移植物抗宿主病的方法。
在一些方面,本文提供用于确定一个或多个基因座(例如,受试者、样品、器官、组织和/或细胞中的基因座)处的等位基因的计算机实现的方法。在一些实施例中,基因座是hla基因座。在一些实施例中,基因座是线粒体高变区(hv)基因座(例如,hv1基因座或hv2基因座)。在一些实施例中,基因座是血型抗原(bga)基因座。在一些实施例中,基因座是中度多态性基因座(即平均每100个核苷酸长度具有至少1个snp的基因座)、高度多态性基因座(即平均每20个核苷酸长度具有至少1个snp的基因座)或极高度多态性基因座(即平均每10个核苷酸长度具有至少1个snp的基因座)。
在一些实施例中,基因座平均每100个碱基含有:1个或更多个但少于20个snp、2个或更多个但少于20个snp、3个或更多个但少于20个snp、4个或更多个但少于20个snp、5个或更多个但少于20个snp、6个或更多个但少于20个snp、7个或更多个但少于20个snp、8个或更多个但少于20个snp、9个或更多个但少于20个snp、10个或更多个但少于20个snp、11个或更多个但少于20个snp、12个或更多个但少于20个snp、13个或更多个但少于20个snp、14个或更多个但少于20个snp、15个或更多个但少于20个snp、16个或更多个但少于20个snp、17个或更多个但少于20个snp、18个或更多个但少于20个snp或19个或更多个但少于20个snp。
在多个实施例中,中度多态性基因座平均每100个碱基含有:1个或更多个但少于5个snp、2个或更多个但少于5个snp、3个或更多个但少于5个snp或4个或更多个但少于5个snp。在多个实施例中,中度多态性基因座平均每100个碱基含有:约1-2个snp、2-3个snp或约3-4个snp。
在多个实施例中,高度多态性基因座平均每100个碱基含有:每100个核苷酸长度5个或更多个但少于10个snp、6个或更多个但少于10个snp、7个或更多个但少于10个snp、8个或更多个但少于10个snp、9个或更多个但少于10个snp。在多个实施例中,高度多态性基因座平均每100个碱基含有:约5-6个snp、约6-7个snp、约7-8个snp或约8-9个snp。
在多个实施例中,极高度多态性基因座平均每100个碱基含有:10个或更多个但少于20个snp、11个或更多个但少于20个snp、12个或更多个但少于20个snp、13个或更多个但少于20个snp、14个或更多个但少于20个snp、15个或更多个但少于20个snp、16个或更多个但少于20个snp、17个或更多个但少于20个snp、18个或更多个但少于20个snp或19个或更多个但少于20个snp。在一个实施例中,极高度多态性基因座平均每100个碱基含有:约10-11个snp、约11-12个snp、约12-13个snp、约13-14个snp、约14-15个snp、约15-16个snp、约16-17个snp、约17-18个snp或约18-19个snp。在一个实施例中,极高度多态性基因座平均每100个碱基含有约20个snp。
在一些实施例中,该计算机实现方法包括:a)在计算机系统上接收序列数据,该序列数据包含多个测序读段;b)通过该计算机系统,将测序读段映射至包含基因座的多个等位基因的参考序列以鉴定候选等位基因;以及c)通过该计算机系统,将最有可能说明映射至基因座的测序读段的候选等位基因对鉴定为存在于该基因座处的等位基因。在一些实施例中,等位基因是hla等位基因、hv等位基因或bga等位基因,并且基因座是hla基因座、hv基因座或bga基因座。在一些实施例中,存在于基因座处的等位基因构成该基因座处的hla类型。在一些实施例中,参考序列还包括基因组序列(例如,基因座被屏蔽或移除的基因组序列)。在一些实施例中,等位基因和序列是人类的。
在一些实施例中,上述方法的步骤b)包括由计算机系统执行的如下步骤:i)将测序读段映射至参考序列,该参考序列包含基因组序列和基因座的多个等位基因序列;ii)将最大数目的测序读段映射至其上的等位基因鉴定为第一组候选等位基因;iii)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因鉴定为第二组候选等位基因;以及iv)如果少于90%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因鉴定为第三组候选等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。术语“蛋白质群组”包括编码具有相同氨基酸序列的同一蛋白质的一组等位基因。在一些实施例中,如果映射至基因座的测序读段(排除映射至第一组候选等位基因的测序读段)大于映射至第一组候选等位基因的测序读段的数目的1%的话,第二组候选等位基因既包括最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因,又包括第二最大数目的测序读段(未排除映射至第一组候选等位基因的测序读段)映射至其上的等位基因。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因的测序读段的数目占映射至基因座的测序读段总数目的至少10%的话,才在步骤iv)中鉴定第三组候选等位基因。
在一些实施例中,上述方法的步骤b)包括由计算机系统执行的如下步骤:i)将测序读段以低严格性映射至参考序列,该参考序列包含人基因组序列和基因座的多个等位基因序列;ii)将来自每个四位数蛋白质家族的所有等位基因鉴定为准候选(pre-candidate)等位基因,所述四位数蛋白质家族的至少一个等位基因属于所映射等位基因的前10%;iii)将测序读段以更高严格性映射至参考序列,该参考序列包含所述准候选等位基因;iv)将最大数目的测序读段映射至其上的准候选等位基因鉴定为第一组候选等位基因;v)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的准候选等位基因鉴定为第二组候选等位基因;以及vi)如果少于90%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的准候选等位基因鉴定为第三组候选等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至基因座的测序读段(排除映射至第一组候选等位基因的测序读段)大于映射至第一组候选等位基因的测序读段的数目的1%的话,第二组候选等位基因既包括最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因,又包括第二最大数目的测序读段(未排除映射至第一组候选等位基因的测序读段)映射至其上的等位基因。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因的测序读段的数目占映射至hla基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。
在一些实施例中,最有可能说明测序读段的候选等位基因对是这样的候选等位基因对,其最有可能说明:i)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);以及ii)存在于映射至候选等位基因的测序读段中的连续的成对snp。一些实施例中,最有可能说明测序读段的候选等位基因对是这样的候选等位基因对,其最有可能说明:i)存在于映射至候选等位基因的测序读段中的独立的snp;ii)存在于映射至候选等位基因的测序读段中的连续snp对;以及iii)候选等位基因对在序列数据的来源生物体中(如人类中)的频率。
在一些实施例中,最有可能说明测序读段的候选等位基因对通过如下方式确定:i)对于每对候选等位基因,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;以及ii)对于每对候选等位基因,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率,其中基因型对数似然分值和相位对数似然分值之和最高的候选等位基因对为最有可能说明测序读段的候选等位基因对。
在一些实施例中,最有可能说明测序读段的候选等位基因对通过如下方式确定:i)对于每对候选等位基因,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;ii)对于每对候选等位基因,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;以及iii)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和,其中基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对为最有可能说明测序读段的候选等位基因对。
在一些方面,本文提供一种计算机实现的方法,该方法包括:a)在计算机系统上接收序列数据,该序列数据包含多个测序读段;b)通过该计算机系统,将测序读段映射至参考序列,参考序列包含基因组序列和基因座的多个等位基因序列;d)通过该计算机系统,将最大数目的测序读段映射至其上的等位基因鉴定为第一组候选等位基因;e)如果少于90%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则通过该计算机系统,将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因鉴定为第三组候选等位基因;f)对于每对候选等位基因,通过该计算机系统,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;g)对于每对候选等位基因,通过该计算机系统,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;h)对于每对候选等位基因,通过该计算机系统,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和;以及i)通过该计算机系统,将基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对鉴定为该基因座处存在的等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至基因座的测序读段(排除映射至第一组候选等位基因的测序读段)大于映射至第一组候选等位基因的测序读段的数目的1%的话,第二组候选等位基因既包括最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因,又包括第二最大数目的测序读段(未排除映射至第一组候选等位基因的测序读段)映射至其上的等位基因。在一些实施例中,等位基因是hla等位基因、hv等位基因或bga等位基因,并且基因座是hla基因座、hv基因座或bga基因座。在一些实施例中,存在于基因座处的等位基因构成该基因座处的hla类型。在一些实施例中,等位基因和序列是人类的。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因的测序读段的数目占映射至基因座的测序读段总数目的至少10%的话,才在步骤e)中鉴定第三组候选等位基因。
在本文提供的计算机实现方法的一些实施例中,序列数据是基因组范围的(genome-wide)测序数据。在一些实施例中,基因组范围的测序数据是转录组测序数据、全外显子组测序数据或全基因组测序数据。在一些实施例中,序列数据的覆盖度低于60倍、50倍、40倍、30倍、20倍或15倍。在一些实施例中,序列数据的覆盖度高于60倍。在一些实施例中,测序读段的平均长度少于100个、90个、80个、70个、60个、50个、45个、40个或35个核苷酸。在一些实施例中,测序读段的长度大于100个核苷酸。
在本文提供的计算机实现方法的某些实施例中,参考序列包括人基因组序列。在一些实施例中,基因组序列中的基因座(例如,hla基因座)的序列已被移除或屏蔽。在一些实施例中,人基因组序列为grch37/hg19。
在一些实施例中,本文所述的方法包括对样品进行基因组范围的测序过程以产生序列数据的步骤。在一些实施例中,本文所述的方法包括执行可产生包含基因座的核酸序列的扩增产物的核酸扩增过程,以及对该扩增产物执行测序过程。
在一些实施例中,本文提供的方法包括向接受者移植hla基因座处的hla类型匹配该受试者在hla基因座处的hla类型的细胞、组织或器官。在一些实施例中,执行本文提供的计算机实现方法来确定接受者在hla基因座处的hla类型。在一些实施例中,执行本文提供的计算机实现方法来确定细胞、组织或器官在hla基因座处的hla类型。在一些实施例中,执行本文提供的计算机实现方法来确定细胞、组织或器官与接受者二者在hla基因座处的hla类型。
在一些方面,本文提供用于执行本文提供的计算机实现方法的计算机系统。在一些实施例中,该计算机系统包括:至少一个处理器;与所述至少一个处理器关联的存储器;显示器;和所述存储器中支持的用于确定基因座处的等位基因(例如,hla基因座处的hla类型)的程序,该程序含有多个指令,当由所述至少一个处理器执行时,这些指令引起所述至少一个处理器执行本文提供的计算机实现方法。在一些实施例中,这些指令在由至少一个处理器执行时,引起所述至少一个处理器:a)接收序列数据,该序列数据包含多个测序读段;b)将测序读段映射至包含基因座的多个等位基因的参考序列以鉴定候选等位基因;以及c)将最有可能说明映射至基因座的测序读段的候选等位基因对鉴定为存在于该基因座处的等位基因。在一些实施例中,这些指令在由至少一个处理器执行时,引起所述至少一个处理器:a)接收序列数据,该序列数据包含多个测序读段;b)将测序读段映射至参考序列,该参考序列包含人基因组序列和基因座的多个等位基因序列;c)将最大数目的测序读段映射至其上的等位基因鉴定为第一组候选等位基因;d)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因鉴定为第二组候选等位基因;e)如果少于90%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因鉴定为第三组候选等位基因;f)对于每对候选等位基因,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;g)对于每对候选等位基因,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;h)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和;以及i)将基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对鉴定为该基因座处存在的等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,等位基因是hla等位基因、hv等位基因或bga等位基因,并且基因座是hla基因座、hv基因座或bga基因座。在一些实施例中,如果映射至基因座的测序读段(排除映射至第一组候选等位基因的测序读段)大于映射至第一组候选等位基因的测序读段的数目的1%的话,第二组候选等位基因既包括最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因,又包括第二最大数目的测序读段(未排除映射至第一组候选等位基因的测序读段)映射至其上的等位基因。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因的测序读段的数目占映射至基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。在一些实施例中,存在于基因座处的等位基因构成该基因座处的hla类型。在一些实施例中,参考序列还包括基因组序列(例如,基因座被屏蔽或移除的基因组序列)。在一些实施例中,等位基因和序列是人类的。
在一些方面,本文提供用于确定基因座处存在的等位基因的计算机程序产品。在一些实施例中,该计算机程序产品存在于非临时性计算机可读介质之上,该介质上存储有多个指令,当由计算机处理器执行时,这些指令引起该计算机处理器执行本文提供的计算机实现方法。在某些实施例中,所述多个指令在由计算机处理器执行时,引起该计算机处理器:a)接收序列数据,该序列数据包含多个测序读段;b)将测序读段映射至包含基因座的多个等位基因的参考序列以鉴定候选等位基因;以及c)将最有可能说明映射至基因座的测序读段的候选等位基因对鉴定为存在于该基因座处的等位基因。在某些实施例中,所述多个指令在由计算机处理器执行时,引起该计算机处理器:a)接收序列数据,该序列数据包含多个测序读段;b)将测序读段映射至参考序列,该参考序列包含人基因组序列和基因座的多个等位基因序列;c)将最大数目的测序读段映射至其上的等位基因鉴定为第一组候选等位基因;d)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因鉴定为第二组候选等位基因;e)如果少于90%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因鉴定为第三组候选等位基因;f)对于每对候选等位基因,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;g)对于每对候选等位基因,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;h)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和;以及i)将基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对鉴定为该基因座处存在的等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至基因座的测序读段(排除映射至第一组候选等位基因的测序读段)大于映射至第一组候选等位基因的测序读段的数目的1%的话,第二组候选等位基因既包括最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因,又包括第二最大数目的测序读段(未排除映射至第一组候选等位基因的测序读段)映射至其上的等位基因。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因的测序读段的数目占映射至基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。
在一些方面,本文提供确定受试者在单倍体dna的基因座(例如,线粒体dna的高变区(hv)基因座)处的基因型。在一些实施例中,该方法包括:a)在计算机系统上接收序列数据,该序列数据包含多个测序读段;b)通过该计算机系统,将测序读段映射至包含基因座的多个等位基因的参考序列以鉴定候选等位基因;以及c)通过该计算机系统,将最有可能说明映射至基因座的测序读段的一个或多个候选等位基因鉴定为存在于该基因座处的等位基因。在一些实施例中,等位基因是hv等位基因,基因座是hv基因座。在一些实施例中,存在于基因座处的等位基因构成该基因座处的基因型。在一些实施例中,参考序列还包括基因组序列(例如,基因座被屏蔽或移除的基因组序列)。在一些实施例中,等位基因和序列是人类的。在一些实施例中,所述方法包括由计算机系统执行的如下步骤:i)将测序读段映射至参考序列,该参考序列包含人基因组序列和基因座的多个等位基因序列;ii)将最大数目的测序读段映射至其上的等位基因鉴定为第一组候选等位基因;iii)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因鉴定为第二组候选等位基因;以及iv)如果少于90%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因鉴定为第三组候选等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果在排除映射至第一组候选等位基因的测序读段之后,映射至基因座的测序读段的数目大于映射至第一组候选等位基因的测序读段的数目的1%,则进一步将第二大数目的未排除映射至第一组候选等位基因的测序读段的测序读段映射至其上的等位基因鉴定为第二组候选等位基因的子集。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因的测序读段的数目占映射至基因座的测序读段总数目的至少10%的话,才在步骤iv)中鉴定第三组候选等位基因。
在一些实施例中,最有可能说明测序读段的一个或多个候选等位基因是这样的一个或多个候选等位基因,其最有可能说明:i)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);以及ii)存在于映射至候选等位基因的测序读段中的连续的成对snp。
在一些实施例中,最有可能说明测序读段的一个或多个候选等位基因是这样的一个或多个候选等位基因,其最有可能说明:i)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);ii)存在于映射至候选等位基因的测序读段中的连续的成对snp;以及iii)该候选等位基因对在人类中的频率。在一些实施例中,最有可能说明测序读段的一个或多个候选等位基因通过如下方式确定:i)对于每个独立的候选等位基因以及每个候选等位基因组合,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述独立的候选等位基因或候选等位基因组合可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;以及ii)对于每个独立的候选等位基因以及每个候选等位基因组合,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述独立的候选等位基因或候选等位基因组合可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率,其中基因型对数似然分值和相位对数似然分值之和最高的独立候选等位基因或候选等位基因组合为最有可能说明测序读段的一个或多个候选等位基因。
在一些实施例中,最有可能说明测序读段的候选等位基因对通过如下方式确定:i)对于每个独立的候选等位基因以及每个候选等位基因组合,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述独立的候选等位基因或候选等位基因组合可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;ii)对于每个独立的候选等位基因以及每个候选等位基因组合,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述独立的候选等位基因或候选等位基因组合可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;以及iii)对于每个独立的候选等位基因以及每个候选等位基因组合,确定频率对数似然分值,频率对数似然分值为每个独立的候选等位基因和每个候选等位基因组合存在于人种群中的频率的对数之和;其中基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的独立候选等位基因或候选等位基因组合为最有可能说明测序读段的一个或多个候选等位基因。
在一些方面,本文提供移植器官、组织或细胞给受试者、防止移植排斥和/或防止移植物抗宿主病的方法。在一些实施例中,该方法包括:a)获得受试者的序列数据,该序列数据包含多个测序读段;b)将测序读段映射至包含hla基因座的多个hla等位基因序列的参考序列以鉴定候选等位基因;c)将最有可能说明映射至hla基因座的测序读段的候选等位基因对鉴定为构成受试者在hla基因座处的hla类型的等位基因;以及d)给受试者移植在hla基因座处的hla类型匹配该受试者在hla基因座处的hla类型的器官、组织或细胞。在一些实施例中,该方法包括:a)获得器官、组织或细胞的序列数据,该序列数据包含多个测序读段;b)将测序读段映射至包含hla基因座的多个hla等位基因序列的参考序列以鉴定候选等位基因;c)将最有可能说明映射至hla基因座的测序读段的候选等位基因对鉴定为构成受试者在hla基因座处的hla类型的等位基因;以及d)将该器官、组织或细胞移植给在hla基因座处的hla类型匹配该器官、组织或细胞在hla基因座处的hla类型的受试者。
在一些实施例中,步骤b)包括如下步骤:i)将测序读段映射至参考序列,该参考序列包含人基因组序列和hla基因座的多个hla等位基因序列;ii)将最大数目的测序读段映射至其上的hla等位基因鉴定为第一组候选等位基因;iii)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的hla等位基因鉴定为第二组候选等位基因;以及iv)如果少于90%的映射至hla基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因鉴定为第三组候选等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因的测序读段的数目占映射至hla基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。
在一些实施例中,步骤b)包括如下步骤:i)将测序读段以低严格性映射至参考序列,该参考序列包含人基因组序列和hla基因座的多个hla等位基因序列;ii)将来自每个四位数蛋白质家族的所有等位基因鉴定为准候选等位基因,所述四位数蛋白质家族的至少一个等位基因属于所映射等位基因的前10%;iii)将测序读段以更高严格性映射至参考序列,该参考序列包含所述准候选等位基因;iv)将最大数目的测序读段映射至其上的准候选等位基因鉴定为第一组候选等位基因;v)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的准候选等位基因鉴定为第二组候选等位基因;以及vi)如果少于90%的映射至hla基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的准候选等位基因鉴定为第三组候选等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因的测序读段的数目占映射至hla基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。
在一些实施例中,最有可能说明测序读段的候选等位基因对是这样的候选等位基因对,其最有可能说明:i)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);以及ii)存在于映射至候选等位基因的测序读段中的连续的成对snp。在一些实施例中,最有可能说明测序读段的候选等位基因对是这样的候选等位基因对,其最有可能说明:i)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);ii)存在于映射至候选等位基因的测序读段中的连续的成对snp;以及iii)该候选等位基因对在人类中的频率。
在一些实施例中,最有可能说明测序读段的候选等位基因对通过如下方式确定:i)对于每对候选等位基因,确定hla基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述hla基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;以及ii)对于每对候选等位基因,确定hla基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述hla基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率,其中基因型对数似然分值和相位对数似然分值之和最高的候选等位基因对为最有可能说明测序读段的候选等位基因对。
在一些实施例中,最有可能说明测序读段的候选等位基因对通过如下方式确定:i)对于每对候选等位基因,确定hla基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述hla基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;ii)对于每对候选等位基因,确定hla基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述hla基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;以及iii)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和,其中基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对为最有可能说明测序读段的候选等位基因对。
在一些方面,移植器官、组织或细胞至受试者、防止移植排斥和/或防止移植物抗宿主病的方法包括:a)获得受试者的序列数据,该序列数据包含多个测序读段;b)将测序读段映射至参考序列,该参考序列包含人基因组序列和hla基因座的多个hla等位基因序列;c)将最大数目的测序读段映射至其上的hla等位基因鉴定为第一组候选等位基因;d)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的hla等位基因鉴定为第二组候选等位基因;e)如果少于90%的映射至hla基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因鉴定为第三组候选等位基因;f)对于每对候选等位基因,确定hla基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述hla基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;g)对于每对候选等位基因,确定hla基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述hla基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;h)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和,其中受试者在该hla基因座处的hla类型为基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对;i)将hla基因座处的hla类型匹配该受试者在hla基因座处的hla类型的器官、组织或细胞移植至受试者。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因的测序读段的数目占映射至hla基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。
在一些实施例中,移植器官、组织或细胞至受试者、防止移植排斥和/或防止移植物抗宿主病的方法包括:a)获得器官、组织或细胞的序列数据,该序列数据包含多个测序读段;b)将测序读段映射至参考序列,该参考序列包含人基因组序列和hla基因座的多个hla等位基因序列;c)将最大数目的测序读段映射至其上的hla等位基因鉴定为第一组候选等位基因;d)将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的hla等位基因鉴定为第二组候选等位基因;e)如果少于90%的映射至hla基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因鉴定为第三组候选等位基因;f)对于每对候选等位基因,确定hla基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述hla基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;g)对于每对候选等位基因,确定hla基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述hla基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;h)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和,其中受试者在该hla基因座处的hla类型为基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对;i)将该器官、组织或细胞移植至hla基因座处的hla类型匹配该器官、组织或细胞在hla基因座处的hla类型的受试者。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。在一些实施例中,如果映射至最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的hla等位基因的测序读段的数目占映射至hla基因座的测序读段总数目的至少10%的话,才鉴定第三组候选等位基因。
在本文提供的方法的一些实施例中,序列数据是基因组范围的测序数据。在一些实施例中,基因组范围的测序数据是转录组测序数据、全外显子组测序数据或全基因组测序数据。在一些实施例中,序列数据的覆盖度低于60倍、50倍、40倍、30倍、20倍或15倍。在一些实施例中,测序读段的平均长度少于100个、90个、80个、70个、60个、50个、45个、40个或35个核苷酸。
在本文提供的方法的某些实施例中,参考序列还包括人基因组序列。在一些实施例中,基因组序列中的hla基因座的序列已被移除或屏蔽。在一些实施例中,人基因组序列为grch37/hg19。
在一些实施例中,本文所述的方法包括对样品进行基因组范围的测序过程以产生序列数据的步骤。在一些实施例中,本文所述的方法包括执行可产生包含hla基因座的核酸序列的扩增产物的核酸扩增过程,以及对该扩增产物执行测序过程。
在本文提供的方法的一些实施例中,器官、组织或细胞包括皮肤、骨头、心脏瓣膜、心脏、肺、肾、肝脏、胰腺、肠、胃、睾丸或它们的部分。在一些实施例中,器官、组织或细胞包括骨髓、造血干细胞或成体干细胞。
附图说明
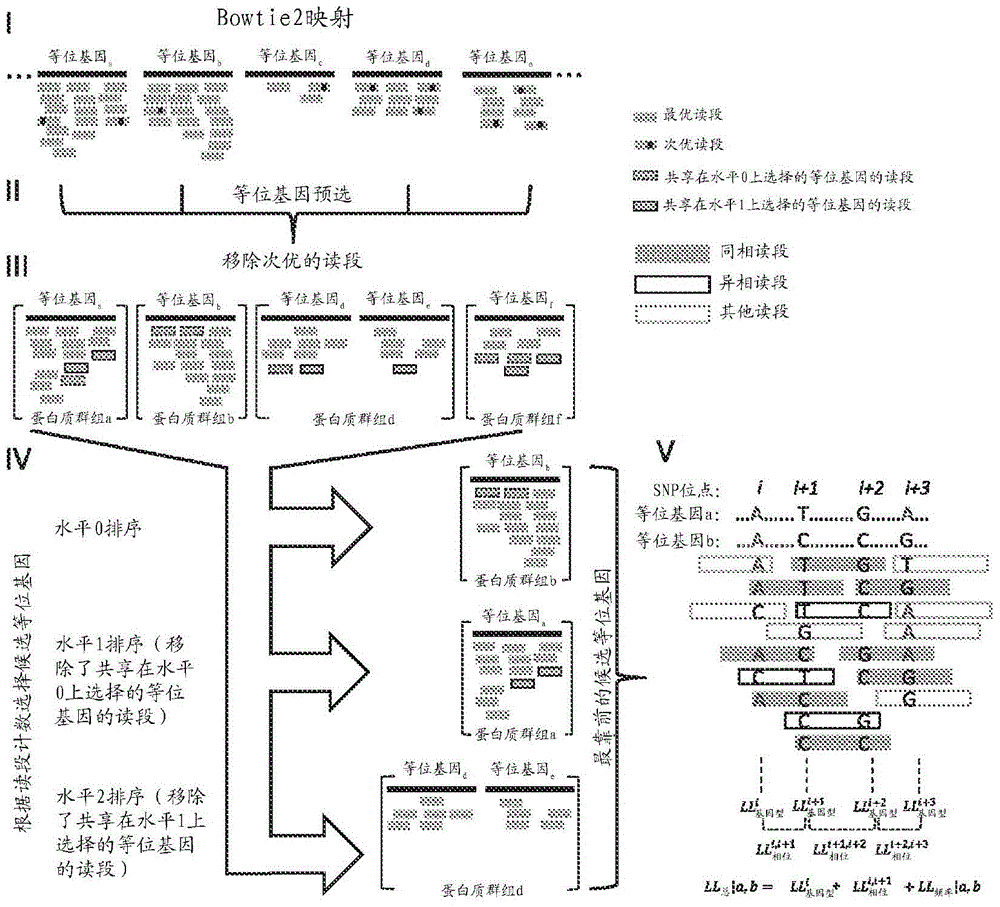
图1为工作流程图,示出了根据一个或多个实施例的示例性方法。该方法包括通过bowtie2将读段映射至hla基因座用独立等位基因的基因组序列替换的人基因组(i)、基于所映射的读段的数目选择靠前的候选等位基因(ii-iv)以及对每对选择的候选等位基因进行对数似然分值评分(v)。
图2为示出了读段长度、覆盖度和测序方案对hla分型精确度的影响的图。该曲线图包括来自hapmaprnaseq数据集的样品(37bp读段长度)、来自genomewxs数据集的样品(100bp长度)和来自hapmapwxs数据集的样品(101bp读段长度)。示出了将输入数据视为双端测序读段(实心符号和实线)和单端测序读段(空心符号和虚线)的预测精确度。符号代表根据其hla基因座的覆盖度倍数分仓(binned)的样品在四位数分辨率下的平均精确度,以误差棒指示方差。就主要i类和ii类hla基因座的cds区计算了映射后的覆盖率倍数,不包括对候选等位基因次优的读段或不与候选等位基因对齐的读段。通过样条内插推导出平滑线以展示符号的趋势。
图3为示出了hapmaprnaseq数据集、1000genomewxs数据集、hapmapwxs和靶向扩增子测序数据集中phlat、hlaminer、hlaforest、seq2hla的预测精确度的表。*对于hapmaprnaseq数据集应用hlaminer的读段比对模式,对于所有其他数据集应用contig组装模式。当计算所有数据集中的seq2hla预测的精确度时未应用p值阈值,这导致与如以前所述施加0.1的p值截止值相比假阴性较低(因而,精确度更高)。#值在早前的出版物的文本中报道。
图4为示出了实例3中用于产生hla序列数据来进行hla分型的靶向扩增子测序策略的示意图。
图5为提供了实例3中用于产生hla序列数据来进行hla分型的靶向扩增子测序策略中所用的引物的表。表格中存在的序列从上到下分别为seqidno:1-22。
图6包括三个图,标记为图(a)、(b)和(c)。图(a)为柱状图,示出了hla-dqa1基因座(左边分图)和hla-dqb1基因座(右边分图)处错误鉴定的等位基因的类型(x轴)和数目(y轴),结果是hapmaprnaseq数据集、1000genomewxs数据集和hapmapwxs数据集的汇总。图(b)为绘出了一个代表性样品中的映射的读段的图表,其中hla-dqa1*03:01等位基因被错误分型为hla-dqa1*03:03等位基因。显示了处于区分这两种等位基因的单snp位置(chr6:32609965,在两条垂直的短划线之间中突出显示)周围的映射读段。hla-dqa1基因的hg19参考序列在该分图的底部示出(seqidno:23)。突出显示的snp的a、c、g、t碱基的累积计数分别是141、117、0和0。图(c)为来自hla-dqa1*03:03等位基因的135核苷酸片段(定为查询片段)与人基因组hg19中的hla-dqa2参考序列的比对。查询序列被简化为平行条棒,仅标出错配。错配处的现有dbsnp记录用红色垂直标志以及相关联的标识号(例如rs62619945)标记,后面跟着指明主要碱基序列和备选碱基序列的括号。框出了将dqa1*03:01等位基因与dqa1*03:03等位基因相区别的snp的比对。图中所示的hla-dqa2的第1-18位核苷酸代表seqidno:24(tcagtcacagaaggtgtt),而其余的核苷酸代表seqidno:25(catt.....ggac)。
图7为工作流程图,示出了根据一个或多个实施例的示例性过程。
图8为工作流程图,示出了根据一个或多个实施例的示例性过程。
具体实施方式
综述
在某些方面,本文提供精确确定基因座(例如高度多态性基因座)处存在的等位基因的方法。在一些实施例中,该方法被称为pat(precisealleletyping,精确等位基因分型)或phlat(precisehlatyping,精确hla分型)。术语phlat和pat可在本文中互换使用。pat方法可广泛适用于鉴定任何基因座(包括高度多态性基因座如hla基因座、bga基因座和hv基因座)处存在的等位基因。pat方法的某些实施例可用于各种各样的应用,包括例如器官移植、个体化用药、诊断学、法医学和人类学。例如,pat方法的实施例可用于防止器官排斥和移植物抗宿主病、用于确定疾病易感性、用于优化疫苗接种策略、用于预测治疗效能以及用于鉴定地理和/或种族起源。
在一些实施例中,pat方法被用于确定hla基因座处的hla类型。pat方法使得能利用各种各样的测序数据,甚至是具有短读段长度和/或低序列覆盖度的测序数据来进行精确的四位数和两位数hla分型。精确的hla类型可基于使用许多不同的测序方法产生的测序数据来预测,这些测序方法包括全基因组范围的测序方法(例如,转录组测序、全外显子组测序和全基因组测序)和hla特异性测序方法(例如,对hla基因座进行核酸扩增,然后对所得的扩增产物进行测序)。
可例如将pat方法用于促进具有匹配或部分匹配的hla类型的供应者和接收者之间的细胞、器官或组织的移植。在一些实施例中,pat方法被用于鉴定和/或促进治疗易发某些疾病或病症的个体,包括免疫原性疾病如狼疮、炎性肠病、多发性硬化症、关节炎和i型糖尿病,以及癌症如乳腺癌或宫颈癌。在一些实施例中,pat方法被用于促进肿瘤免疫疗法和/或癌疫苗接种疗法。在某些实施例中,pat方法被用于确定受试者或样品的地理和/或种族起源。
在某些实施例中,pat方法包括两个部分:1)从基因座的可能等位基因中选择候选等位基因;以及2)对候选等位基因对进行排序以鉴定哪个候选等位基因对最有可能是基因座处的等位基因对。在一些实施例中,基于读段计数来选择候选等位基因。在一些实施例中,基于观察到的数据可由每个等位基因对来说明的可能性来对候选等位基因对进行排序。在一些实施例中,基于独立位置处的序列一致性和连续位置上的相位一致性二者来确定最有可能的等位基因。在一些实施例中,等位基因在人种群中的频率也被纳入到等位基因对的排序中。示出了根据一个或多个实施例的示例性pat方法的流程图在图7和图8中提供。
在一些实施例中,本文所述的方法可用于确定任何主要或次要hla基因座的hla类型。在一些实施例中,hla基因座为i类hla基因座。在一些实施例中,hla基因座为hla-a基因座、hla-b基因座或hla-c基因座。在一些实施例中,hla基因座为ii类hla基因座。在一些实施例中,hla基因座为hla-dqa1基因座、hla-dqb1基因座、hla-dra基因座、hla-drb1基因座、hla-drb3基因座、hla-drb4基因座、hla-drb5基因座、hla-dpa1基因座或hla-dpb1基因座。在一些实施例中,hla基因座为次要hla基因座。hla等位基因的序列是本领域已知的。例如,hla等位基因的基因组序列和编码dna序列(cds)可从imgt的3.8.0版本获得。
在一些实施例中,本文所述的方法用于确定线粒体dna基因座,例如hv基因座(例如,高变区1(hv1)基因座或高变区2(hv2)基因座)的基因型。不像细胞核dna(其为二倍体,因而具有两个拷贝的每种基因座),线粒体dna为单倍体,因而,理论上,将仅含有一个拷贝的基因座。然而,线粒体dna中的基因座通常加倍。因而有可能线粒体dna含有一个、两个或多个拷贝的基因座。因而,当本文所述的方法应用于线粒体dna(或任何由单倍体基因组(包括胚细胞基因组、病毒基因组或细菌基因组)编码的基因座)时,一个或多个等位基因将被鉴定为存在于基因座处,而不是一对等位基因。hv等位基因的序列是本领域已知的。hv等位基因序列可例如在hvrbase++数据库(http://www.hvrbase.org)中找到,如kohl等人,nucleicacidsresearch(《核酸研究》)34:d700-d704(2006)中所述,特此将该文献以引用的方式全文并入。
在一些实施例中,本文所述的方法用于确定bga基因座处存在的等位基因。示例性的bga基因座包括abo基因座和rh基因座。bga基因座等位基因的序列是本领域已知的。例如,bga基因座序列可从ncbesbloodgroupantigengenemutation(ncbes血型抗原基因突变)数据库(http://www.ncbi.nlm.nih.gov/projects/gv/rbc/xslcgi.fcgi?cmd=bgmut)获得,如patnaik等人,nucleicacidsresearch40:d1023-d1029(2012)中所述,特此将该文献以引用的方式全文并入。
在某些实施例中,本文描述的方法是计算机实现的。该方法可在软件、硬件、固件或它们的任何组合中实现。该方法优选在一个或多个计算机程序中实现,该计算机程序在可编程计算机系统上执行,该计算机系统包括至少一个处理器、可由所述处理器读取的存储介质(包括,例如易失性和非易失性记忆和/或存储元件),以及输入和输出设备。该计算机系统可包括一个或多个实体机或者在一个或多个实体机上运行的一个或多个虚拟机。此外,该计算机系统可包括计算机集群或者通过互联网或其他网络连接的多个分散的计算机。
每个计算机程序可以是存在于该计算机系统的随机存取存储器中的代码模块中的一套指令或程序代码。在计算机系统需要之前,该套指令可储存在另一计算机储存器中(例如,硬盘驱动器中,或移动存储器如光盘、外置硬盘、记忆卡或闪存驱动器中)或存储在另一计算机系统上并通过互联网或其他网络下载。每个计算机程序可以各种计算机编程语言(包括,例如python)来实现。
测序数据
在某些实施例中,本文所公开的方法包括获得或接收序列数据的步骤(图7和图8的步骤10)。在一些实施例中,可通过任何方法获得或接收序列数据。例如,可通过对样品执行测序程序来直接获得序列数据。或者,可例如从第三方、数据库和/或出版物间接获得序列数据。在一些实施例中,在计算机系统处接收序列数据,例如从数据存储设备或从一单独的计算机系统接收数据。
本文所述的方法能够使用各种各样的序列数据精确地预测基因座处存在的等位基因(例如,基因座的hla类型)。例如,在一些实施例中,序列数据为基因组范围的测序数据。在一些实施例中,序列数据为转录组测序数据。在一些实施例中,序列数据是全外显子测序数据。在一些实施例中,测序数据为全基因组测序数据。在一些实施例中,序列数据富集编码所述基因座的序列数据。在一些实施例中,序列数据为rna序列数据。在一些实施例中,序列数据为dna序列数据。
在一些实施例中,序列数据包含多个测序读段。在一些实施例中,测序读段的平均读段长度不超过35、36、37、38、39、40、45、50、55、60、65、70、75、80、85、90、95、100、125、150、175、200、250、300、400、500、600、700、800、900或1000个核苷酸。在一些实施例中,测序读段的平均读段长度为至少30、31、32、33、34、35、36、37、38、39、40、45、50、55、60、65、70、75、80、85、90、95、100、125、150、175、200或250个核苷酸。在一些实施例中,测序读段的覆盖度不超过100倍、90倍、80倍、70倍、60倍、50倍、40倍、30倍或20倍。在一些实施例中,测序读段的覆盖度为至少50倍、45倍、40倍、35倍、30倍、25倍、20倍、19倍、18倍、17倍、16倍、15倍、14倍、13倍、12倍、11倍或10倍。
在一些实施例中,可通过本领域已知的任何测序方法产生序列数据。例如,在一些实施例中,利用如下方法产生测序数据:链终止测序法、连接测序法、边合成边测序法、焦磷酸测序法、离子半导体测序法、单分子实时测序法、dilute-‘n’-go测序法和/或454测序法。
在一些实施例中,序列数据是这样一种方法的结果:凭借该方法,进行核酸扩增过程来扩增一个或多个基因座或转录物的至少一部分,然后对所得的扩增产物进行测序。可用于执行本文所公开的方法的核酸扩增过程包括但不限于:聚合酶链反应(pcr)、late-pcr、连接酶链反应(lcr)、链置换扩增(sda)、转录介导的扩增(tma)、自主序列复制(3sr)、基于qβ复制酶的扩增、基于核酸序列的扩增(nasba)、修复链反应(rcr)、自返式dna扩增(boomerangdnaamplification,bda)和/或滚环扩增(rca)。
在一些实施例中,该方法包括对样品执行测序过程的步骤。可以使用任何样品,只要该样品含有dna和/或rna(例如编码hla分子的dna或rna)。在一些实施例中,样品来自有前景的器官、细胞或组织供应者。在一些实施例中,样品来自有前景的器官、细胞或组织接受者。样品来源可以是例如实体组织,如来自新鲜、冷冻和/或保存的器官、组织样品、活检样品或抽吸物;血液或任何血液成分、血清、血液;体液如脑脊液、羊水、腹膜液或间质液、尿液、唾液、粪便、眼泪;或来自受试者怀孕或发育中的任何时间的细胞。
在一些实施例中,执行本领域可用的任何测序方法。在一些实施例中,利用如下方法执行测序:链终止测序法、连接测序法、边合成边测序法、焦磷酸测序法、离子半导体测序法、单分子实时测序法、dilute-‘n’-go测序法和/或454测序法。在一些实施例中,执行核酸扩增过程来扩增一个或多个基因组基因座或转录物(例如,hla基因组基因座或转录物)的至少一部分,然后对所得的扩增产物进行测序。在一些实施例中,所执行的核酸扩增方法为聚合酶链反应(pcr)、late-pcr、连接酶链反应(lcr)、链置换扩增(sda)、转录介导的扩增(tma)、自主序列复制(3sr)、基于qβ复制酶的扩增、基于核酸序列的扩增(nasba)、修复链反应(rcr)、自返式dna扩增(boomerangdnaamplification,bda)和/或滚环扩增(rca)。
候选等位基因的选择
在一些实施例中,本文所公开的方法包括选择候选等位基因的步骤(例如,图7的步骤20和30以及图8的步骤20、32、34和36)。在一些实施例中,候选等位基因的选择通过这样来执行:将测序读段映射至参考序列,然后进行一系列读段计数步骤。该映射过程可例如使用任何可用的序列映射软件来执行。在某些实施例中,使用bowtie2。在一些实施例中,bowtie2映射参数设定为very-sensitive(十分灵敏)(即-d20-r3-n0-l20-is,1,0.50)、end-to-end(端对端)模式,在一些实施例中,参考序列包括多个等位基因,如hla等位基因(例如,在人工染色体上)。在一些实施例中,参考序列还包括人基因组序列(例如,grch37/hg19)。在一些实施例中,将人基因组序列中的一个或多个基因座(例如,hla基因座)从参考序列中排除或将其屏蔽(例如,通过用ns替换该基因座序列)。
参考序列中所包括的等位基因可从等位基因序列的任何来源获得。例如,如果hla等位基因包括在参考序列中,则可从imgt版本3.8.0获得该等位基因的基因组dna序列和编码dna序列(cds)并将其映射至人参考基因组版本37/hg19中的坐标上。在一些实施例中,仅将从转录起始位点到终止密码子的等位基因的基因组序列包括在参考序列中。可通过用参考等位基因的基因组序列(例如,来自hg19基因组的对应基因座处的序列)填充进非编码区中,来使用仅具有cds而无基因组记录(genomicrecord)的等位基因。不受理论的束缚,非编码序列的基因组序列填补(imputation)对hla分型具有极小或没有影响,因为非编码区中的多态性不会在蛋白质水平上改变hla类型。
在一些实施例中,在选择候选等位基因之前,通过以低严格性将序列读段映射至参考序列来选择准候选等位基因(pre-candidateallele)。在一些实施例中,将读段计数的上分位数阈值(例如上侧第95、第90、第85、第80、第75、第70、第65、第60、第55或第50百分位数)应用于对可能的等位基因的粗预选。在一些实施例中,上分位数阈值为上侧第90百分位数。在一些实施例中,上分位数阈值为第70百分位数。在一些实施例中,如果在基因座处存在大量等位基因(例如,至少200、300、400、500、600、700、800、900或1000个等位基因)则上分位数为上侧第90百分位数,但是如果少量等位基因存在于基因座处(例如,不超过200、300、400、500、600、700、800、900或1000个等位基因)则上分位数阈值为上侧第70百分位数。在一些实施例中,只要一蛋白质(四位数)家族的至少一个成员落入在阈值内就保留来自该家族的所有等位基因。在某些实施例中,将来自其至少一个等位基因属于映射的等位基因的前5%、10%、15%、20%、25%或30%的每个四位数蛋白质家族的所有等位基因选择为准候选等位基因。在一些实施例中,选择映射的等位基因的前10%。在一些实施例中,选择映射的等位基因的前30%。在一些实施例中,如果基因座处存在大量等位基因(例如,至少200、300、400、500、600、700、800、900或1000个等位基因)则选择映射的等位基因的前10%,但是如果少量等位基因存在于基因座处(例如,不超过200、300、400、500、600、700、800、900或1000个等位基因)则选择映射的等位基因的前30%。在一些实施例中,仅将准候选等位基因包括在后续的候选选择过程中。在一些实施例中,参考序列中的所有等位基因均包括在后续的候选选择过程中。该预选过程的一个示例性实施例在图1的步骤i和ii中示出。
在一些实施例中,使用严格的标准计算映射到保留的等位基因的读段的数目。例如,在一些实施例中,仅为读段最匹配的等位基因(或多个等位基因,如果联系在一起的话)对读段进行计数,所述匹配通过相应基因座内被读段所覆盖的snp位点上的序列同一性来判断。在一些实施例中,要求至少99%的序列同一性才能对读段进行计数。在一些实施例中,每个基因座的snp为该基因座处保留的等位基因的多态性位点。在一些实施例中,将与任何保留的等位基因中的插入和缺失相重合的位点排除在外。该映射过程的一个示例性实施例在图1的步骤iii中示出。
在某些实施例中,使用一系列读段计数步骤(图8的步骤32、34和36)选择候选等位基因。在一些实施例中,将最大数目的测序读段映射至其上的等位基因鉴定为第一组候选等位基因。在一些实施例中,将最大数目的除了映射至第一组候选等位基因的测序读段之外的测序读段映射至其上的等位基因鉴定为第二组候选等位基因。在一些实施例中,如果少于95%、90%、85%或80%的映射至基因座的测序读段映射至第一组或第二组候选等位基因的等位基因,则将最大数目的除了映射至第一组或第二组候选等位基因的读段之外的测序读段映射至其上的等位基因鉴定为第三组候选等位基因。在一些实施例中,鉴定的等位基因选自一组蛋白质群组。
候选等位基因选择过程的一个示例性实施例在图1的步骤iv中示出。在该实施例中,首先将等位基因根据读段计数从高到低排序(在图1中称为水平0排序)。选择具有最大读段计数的等位基因(或多个等位基因,如果联系在一起的话)并储存为候选等位基因。然后通过排除共享之前选择的等位基因的读段,来调整剩余等位基因中的读段计数。将调整后的读段计数以降序排序(在图1中称为水平1排序)并将新的最靠前的等位基因(或者多个等位基因,如果联系在一起的话)选择为候选等位基因。为了容忍读段映射和计数中的不确定性,将来自水平0上排序第二靠前的等位基因的等位基因作为候选等位基因包括在内,条件是如果它们具有不可忽略数目的不同于最靠前等位基因的读段的话。例如,在一些实施例中,将在排除映射至第一组候选等位基因的测序读段之前第二最大数目的测序读段映射至其上的等位基因包括在水平1排序中,条件是如果在排除映射至在水平0排序上选择的等位基因的读段之后,它们保留的测序读段的数目为映射至水平0排序的等位基因的测序读段的数目的1%的话。如果选自水平0和水平1排序的等位基因说明不到90%的映射至基因座的等位基因的话,则重复该读段计数程序(在图1中称为水平2排序)并将新的最靠前的等位基因(或多个等位基因,如果联系在一起的话)包括在候选等位基因中,条件是如果至少10%的映射至基因座的测序读段映射至新的最靠前的一个等位基因或多个等位基因的话。
在一些实施例中,如果满足以下标准的话则将基因座确定为纯合的(即基因座的两个拷贝均含同一等位基因):在水平0上最靠前等位基因说明至少80%、85%、90%或95%的读段,并且没有其他等位基因说明超过3%、4%、5%、6%、7%、8%、9%或10%的剩余读段。在一些实施例中,如果满足以下标准的话则将基因座确定为纯合的:在水平0上的最靠前等位基因说明至少90%的映射至基因座的读段,并且没有其他等位基因说明超过5%的映射至基因座的读段,映射至在水平0上最靠前的等位基因的读段排除在外。
可能性排序
在某些实施例中,在执行上述候选等位基因选择过程之后,仅将候选等位基因及它们相关的读段包括在后续的分析中。在一些实施例中,将候选等位基因在候选等位基因的所有成对组合(包括自身的对)上进行评价,以发现最有可能存在于基因座处的对(例如,最有可能构成hla类型的对)。本方法的该方面的例子在图7的步骤40以及图8的步骤42、44和46中示出。
在一些实施例中,本文所提供的方法包括鉴定最有可能为存在于基因座处的等位基因的候选等位基因对的步骤。在一些实施例中,所鉴定的候选等位基因对为最有可能说明映射至基因座的测序读段的序列的对。在一些实施例中,所鉴定的候选等位基因对是这样的对,其最有可能说明:1)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);以及2)存在于映射至候选等位基因的测序读段中的连续的成对snp。在一些实施例中,所鉴定的候选等位基因对是这样的对,其最有可能说明:1)存在于映射至候选等位基因的测序读段中的独立的单核苷酸多态性(snp);2)存在于映射至候选等位基因的测序读段中的连续的成对snp;以及3)候选等位基因对在人类中的频率。
在一些实施例中,最有可能说明映射至候选等位基因的测序读段的序列的候选等位基因对通过如下方式确定:1)对于每对候选等位基因,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;以及2)对于每对候选等位基因,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;其中基因型对数似然分值和相位对数似然分值之和最高的候选等位基因对为最有可能说明测序读段的候选等位基因对。
在一些实施例中,最有可能说明映射至候选等位基因的测序读段的序列的候选等位基因对通过如下方式确定:1)对于每对候选等位基因,确定基因座中的每个独立snp的基因型对数似然分值,每个基因型对数似然分值为所述基因座中的每个独立snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述snp的测序读段中的独立snp处存在的序列的对数概率;2)对于每对候选等位基因,确定基因座中每个连续的成对snp的相位对数似然分值,每个相位对数似然分值为所述基因座中的每个连续的成对snp的对数概率之和,所述对数概率为所述候选等位基因对可以说明映射至所述连续的成对snp的测序读段中的连续成对snp处存在的序列的对数概率;以及3)对于每对候选等位基因,确定频率对数似然分值,频率对数似然分值为候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数之和;其中基因型对数似然分值、相位对数似然分值和频率对数似然分值之和最高的候选等位基因对为最有可能说明测序读段的候选等位基因对。
在一些实施例中,将具有最高对数似然分值(ll总)的候选等位基因对鉴定为基因座处存在的等位基因(例如,hla基因座处的hla类型)。在一些实施例中,根据公式1计算ll总。如公式1中所示,每个等位基因对的(ll总)整合了单独snp位点的所观察到的基因型的似然性(ll基因型)和多个位点上的相位的似然性(ll相位),还有等位基因对存在于人类中概率(ll频率)。
基因型似然分值评分
在一些实施例中,根据贝叶斯模型计算基因座中的单独snp的对数似然分值(ll基因型)。在一些实施例中,后验对数似然值lli基因型与条件对数似然值logp(di│gi)成比例,条件对数似然值为观察到累积的碱基(di)给出位点i处的所关注等位基因对(gi)的基因型的对数概率。对于任何基因型假定边缘先验logp(gi)是恒定的,因而将其除去。p(di│gi)为在位点i处观察到碱基j的独立条件对数似然值p(bij│gi)的乘积(公式2)。
qj为从碱基j的phred分值转化而来的错误率。
相位似然分值评分
在一些实施例中,类似于上述的一个snp位点的基因型似然值,对两个相邻snp位点的相位似然值(ll相位)进行建模,
公式3避免了偏好具有非均相序列的等位基因对
等位基因频率评分
在一些实施例中,当确定最有可能的候选等位基因对时,考虑了候选等位基因对中的每个候选等位基因存在于人种群中的频率的对数。主要的i类和ii类基因座的等位基因频率是本领域已知的。例如,这种等位基因频率可从allelefrequencynet下载。在一些实施例中,对于每个蛋白质(四位数)家族,使用了来自文献发表的等位基因的最大频率并为其中的所有等位基因共有。在一些实施例中,将背景值0.0001赋给任何具有未知频率的蛋白质家族(及其等位基因)。在一些实施例中,llfreq被计算为两个等位基因的频率的对数之和。
移植方法
在一些方面,本文所描述的hla分型方法可用于降低移植排斥和/或移植物抗宿主病的可能性。在一些方面,本文提供执行器官、细胞或组织移植的方法。在一些实施例中,移植方法包括:执行本文所述的hla分型方法以确定器官、组织或细胞在至少一个hla基因座处的hla类型,然后将所述器官、组织或细胞移植给接受者。在一些实施例中,移植方法包括:执行本文所述的hla分型方法以确定有前景的移植接受者在至少一个hla基因座处的hla类型,然后将器官、组织或细胞移植给所述接受者。在一些实施例中,移植方法包括:执行本文所述的hla分型方法以确定器官、组织或细胞在至少一个hla基因座处的hla类型,执行本文所述的hla分型方法以确定有前景的移植接受者在至少一个hla基因座处的hla类型,然后将所述器官、组织或细胞移植给所述接受者。
在一些方面,本文提供防止移植的器官、组织或细胞排斥的方法。在一些实施例中,该方法包括:执行本文所述的hla分型方法以确定器官、组织或细胞在至少一个hla基因座处的hla类型,然后将所述器官、组织或细胞移植给接受者。在一些实施例中,该方法包括:执行本文所述的hla分型方法以确定有前景的移植接受者在至少一个hla基因座处的hla类型,然后将器官、组织或细胞移植给所述接受者。在一些实施例中,该方法包括:执行本文所述的hla分型方法以确定器官、组织或细胞在至少一个hla基因座处的hla类型,执行本文所述的hla分型方法以确定有前景的移植接受者在至少一个hla基因座处的hla类型,然后将所述器官、组织或细胞移植给所述接受者。
在一些方面,本文提供防止移植物抗宿主病的方法。在一些实施例中,该方法包括:执行本文所述的hla分型方法以确定器官、组织或细胞在至少一个hla基因座处的hla类型,然后将所述器官、组织或细胞移植给接受者。在一些实施例中,该方法包括:执行本文所述的hla分型方法以确定有前景的移植接受者在至少一个hla基因座处的hla类型,然后将器官、组织或细胞移植给所述接受者。在一些实施例中,该方法包括:执行本文所述的hla分型方法以确定器官、组织或细胞在至少一个hla基因座处的hla类型,执行本文所述的hla分型方法以确定有前景的移植接受者在至少一个hla基因座处的hla类型,然后将所述器官、组织或细胞移植给所述接受者。在一些实施例中,以2位数分辨率确定hla类型。在一些实施例中,以4位数分辨率确定hla类型。
在一些实施例中,在移植之前测试的hla基因座是i类hla基因座。在一些实施例中,hla基因座为hla-a基因座、hla-b基因座或hla-c基因座。在一些实施例中,hla基因座为ii类hla基因座。在一些实施例中,hla基因座为hla-dqa1基因座、hla-dqb1基因座、hla-dra基因座、hla-drb1基因座、hla-drb3基因座、hla-drb4基因座、hla-drb5基因座、hla-dpa1基因座或hla-dpb1基因座。在一些实施例中,确定多种hla基因座的hla类型。例如,在一些实施例中,确定至少2、3、4、5、6、7、8、9、10、11或12种hla基因座的hla类型。在一些实施例中,确定所有三种i类hla基因座(hla-a、hla-b和hla-c)的hla类型。在一些实施例中,确定hla-a、hla-b、hla-c、hla-dqa1、hla-dqb1和hla-drb1的hla类型。在一些实施例中,确定hla-a、hla-b和hla-drb1的hla类型。
在一些实施例中,器官、组织或细胞的hla类型匹配接受者在hla基因座处的hla类型。在一些实施例中,hla基因座为hla-a基因座、hla-b基因座或hla-c基因座。在一些实施例中,hla基因座为hla-dqa1基因座、hla-dqb1基因座、hla-dra基因座、hla-drb1基因座、hla-drb3基因座、hla-drb4基因座、hla-drb5基因座、hla-dpa1基因座或hla-dpb1基因座。在一些实施例中,器官、组织或细胞的hla类型匹配接受者在至少2、3、4、5、6、7、8、9、10、11或12种hla基因座处的hla类型。在一些实施例中,器官、组织或细胞的hla类型匹配接受者在至少2种i类hla基因座处的hla类型。在一些实施例中,器官、组织或细胞的hla类型匹配接受者在全部3种i类hla基因座处的hla类型。在一些实施例中,器官、组织或细胞的hla类型匹配接受者在hla-a基因座和hla-b基因座处的hla类型。在一些实施例中,器官、组织或细胞的hla类型匹配接受者在hla-a基因座、hla-b基因座和hla-drb1基因座处的hla类型。在一些实施例中,器官、组织或细胞的hla类型不匹配接受者在不超过11、10、9、8、7、6、5、4、3、2或1种hla基因座处的hla类型。在一些实施例中,该匹配具有2位数分辨率。在一些实施例中,该匹配具有4位数分辨率。
在本文提供的方法的一些实施例中,进行器官移植。在一些实施例中,所移植的器官是心脏、肺、肾、肝脏、胰腺、肠、胃和/或睾丸或者前述器官其中一者的一部分。在一些实施例中,所移植的细胞、组织或器官是肢体(例如,手、脚、手臂或腿)、角膜、皮肤、脸、胰岛、骨髓、造血干细胞、成体干细胞(例如,乳腺干细胞、肠干细胞、间充质干细胞、内皮干细胞、神经干细胞、嗅觉干细胞、心干细胞、肺干细胞)、血管、心脏瓣膜和/或骨。移植的器官、组织或细胞可来自活的供应者或已故供应者。
在本文提供的方法的一些实施例中,给所述器官、组织或细胞的接受者施用可降低移植排斥可能性的药剂。在一些实施例中,该药剂是免疫抑制剂。在某些实施例中,给接受者施用泼尼松龙、氢化可的松、环孢素、他克莫司、咪唑硫嘌呤、霉酚酸、西罗莫司、依维莫司、巴利昔单抗、达珠单抗、抗胸腺细胞球蛋白、抗淋巴细胞球蛋白和/或利妥昔单抗。在一些实施例中,如果接受者的hla类型不匹配移植的器官、细胞或组织在一个或多个hla基因座处的hla类型,则给接受者施用该药剂。在一些实施例中,如果接受者的hla类型不匹配移植的器官、细胞或组织在至少1、2、3、4、5、6、7、8、9、10或11个hla基因座处的hla类型,则给接受者施用该药剂。
特此将本文提及的所有出版物(包括专利、专利申请以及genbank登录号)以引用的方式全文并入,就如同明确且独立地指明将每篇单独的出版物或专利以引用的方式并入一样。在发生冲突的情况下,以本专利申请(包括本文中的任何定义)为准。
现在本发明得到了一般性的描述,通过参考下面的实例将更容易理解本发明,将这些实例包括在内仅仅是为了示例说明本发明的某些方面和实施例,并非旨在限制本发明。
实例
实例1:使用phlat方法的实施例进行hla分型
phlat工作流程从使用bowtie2进行基于参考序列的读段映射(图1中的步骤i)开始。通过用一批人工染色体扩展人基因组grch37/hg19来构建参考基因组,该批人工染色体每一者提供一个hla等位基因的基因组dna序列。通过n’s屏蔽染色体6上的hla-a、b、c、dqa1、dqb1和drb1基因座处的对应基因组序列,以避免双重映射。bowtie2映射参数设定为:very-sensitive(十分灵敏)(即-d20-r3-n0-l20-is,1,0.50),—end-to-end(端对端)模式。报告每个读段的最佳比对(或同样好的比对中的其中之一)。当读段长度适用于bowtie时,将映射引擎换成bowtie没有明显改变phlat的性能(未示出数据)。
主要的i类和ii类基因座hla-a(1884个)、hla-b(2489个)、hla-c(1382个)、hla-dqa1(47个)、hla-dqb1(165个)和hla-drb1(1092个)的总共7059个等位基因被包括在该参考序列中。所述等位基因的基因组dna序列和编码dna序列(cds)从imgt版本3.8.0获得并映射至人参考基因组版本37/hg19中的坐标。基因组dna序列用于bowtie2映射(图1,步骤i,见下文),而cds序列用于所有其他程序(图1步骤ii-v)。仅保留从转录起始位点(tss)至终止密码子的基因组序列。对于任何仅具有cds而无基因组记录的等位基因,将非编码区用hg19基因组中相应基因座处使用的参考等位基因(例如,a*03:01:01:01为hla-a基因座的参考等位基因)的基因组序列填充,只要没有可用的数据表明有超出该等位基因的cds区的变异。基因组序列填补对hla分型具有极小(如果有的话)影响,因为非编码区中的多态性不会在蛋白质水平上改变hla类型。
后面的hla类型预测在两个主要的步骤中完成:靠前候选等位基因的选择(图1中的步骤ii-iv)以及基于似然值的排序(图1中的步骤v)。该等位基因选择大大减少了似然值排序的计算成本,因为在似然值排序过程中必须评价等位基因的每个成对组合。随后,似然分值整合了基因型和相位信息以及先验知识来以高分辨率解析高度同源的hla等位基因。
靠前的候选等位基因的选择涉及读段计数的迭代。首先,根据bowtie2映射结果,对映射至每个等位基因的读段数目进行计数。将读段计数的上分位数阈值(如第90百分位数)应用于对可能的等位基因的粗略预选(图1中的步骤ii)。只要一个肽(四位数)家族的一个成员被选择,就保留来自该家族的所有等位基因。接下来,根据更严格的标准重新计算映射至保留的等位基因的读段的数目(图1中的步骤iii)。使用由bowtie2输出的每个读段的坐标,将读段与该位置处所有保留的等位基因进行比较。仅对与读段匹配最好的等位基因(或多个等位基因,如果联系在一起的话)的该读段进行计数,这通过在相应基因座内由该读段覆盖的snp位点上的序列同一性来判断。要求至少99%的序列同一性才能对读段进行计数。每个基因座的snp为来自该基因座处保留的等位基因的多态性位点的集合。将与任何保留的等位基因中的插入和缺失重合的位点排除在外以避免比对偏差,因为插入和缺失未被视为不匹配。根据蛋白质群组(四位数)将读段计数进行非冗余汇总,并用于通过连续的基于计数的排序进行靠前候选等位基因的选择(图1的步骤iv)。具体而言,对于给定的基因座,首先通过读段计数从高到低对蛋白质群组进行排序,称为水平0排序。选择具有最大读段计数的群组(或多个群组,如果联系在一起的话),并将所有关联的等位基因作为候选等位基因储存。然后通过将共享前面选择的群组的读段排除在外,来调整剩余蛋白质群组中的读段计数。将调整过的读段计数以降序排序(水平1排序)并选择新的最靠前群组。为了容忍读段映射和计数中的不确定性,尤其是当测序覆盖度有限或真等位基因或假等位基因十分类似时,将来自在水平0上排序第二靠前的蛋白质群组的等位基因包括在内,条件是如果它们具有不可忽略数目的未共享所述最靠前群组的独特读段(大于映射至排序最靠前的群组的读段的1%)的话。通常选自水平0和水平1排序的等位基因可以解释大部分(≥90%)映射至基因座的读段。否则,重复该程序(水平2排序)并选择基因座处的新的最靠前蛋白质群组。
如果满足以下标准的话,则可在该候选等位基因选择步骤确定四位数分辨率的纯合基因型:在水平0上最靠前蛋白质群组解释大部分的读段(>90%),并且与已解释的读段相比由任何其他群组解释的其余读段可忽略不计(少于5%)。
在该选择结束时,仅将候选等位基因及它们的相关读段用于后续分析。通常,保留数十个等位基因。该数目小得足以在所述等位基因的全部成对组合(包括自身组合)上进行穷举评价以发现最有可能的对。如公式1中所示,每个等位基因对的总对数似然分值(ll总)整合了所观察到的基因型在单独snp位点上的似然性(ll基因型)和多个位点上的相位似然性(ll相位),还有等位基因对存在于人类中概率(ll频率)。
基于贝叶斯模型,后验对数似然值lli基因型与条件对数似然值log.p(di│gi)成比例,条件对数似然值为观察到累积的碱基(di)给出位点i处的所关注等位基因对(gt)的基因型的对数概率。对于任何基因型假定边缘先验logp(gi)是恒定的,因而将其除去。p(di│gi)为在位点i观察到碱基j的独立条件对数似然值
qj为从碱基j的phred分值转化而来的错误率。
类似于一个snp位点的基因型似然值,对两个相邻snp位点的相位似然值进行建模。在两个位点的情况下,存在15个可能的错配(异相)状态和1个匹配(同相状态),而不是对于单个位点而言的3个错配和1个匹配。具体而言,
图3避免了先前的工作中的偏好具有非均相序列的等位基因对
主要的i类和ii类基因座的等位基因频率从allelefrequencynet下载。对于每个蛋白质(四位数)家族,使用了来自文献发表的等位基因的最大频率并为其中的所有等位基因共有。将背景值0.0001赋给具有未知频率的蛋白质家族(及其等位基因)。ll频率被计算为两个等位基因的频率的对数之和。
将具有最高ll总的等位基因对报告为预测的hla类型。通常,ll总由ll基因型分量和ll相位分量决定。ll频率显著较小,通常小数个数量级。因而,尽管应用的等位基因频率可能存在不确定性,但我们预计对结果没有明显的影响。
实例2:phlat使用短读段精确地确定hla类型
为了评价使用短读段的phlat,使用了hapmap转录组测序(rnaseq)数据集。使用双端短读段(2×37bp)进行的类淋巴母细胞的转录组剖析是从来自hapmap项目(研究登录号为erp000101)的公用数据库获得,该数据库是祖先来自北欧和西欧的60位犹他州居民的数据库。最初由debakker等人nat.genet.(《自然遗传学》)38:1166-1172(2006)以四位数分辨率在主要的i类和ii类hla基因座处对五十份这些样品进行基因型分型,并随后在erlich等人,bmcgenomics(《bmc基因组学》)12:42(2011)中使用不同的技术进行了验证。一份样品(轮次登录号(runaccession)err009139)由于可映射至人基因组的读段的比率异常的低(<20%)而被排除。将剩余的49个受试者用于本工作中的分析和比较。
hapmaprnaseq数据采用双端37bp读段。类似的读段长度(约35bp)通常用于转录组测序研究。然而,它们处于适用的读段长度的下限之内。采用现有技术,使用这样十分短的读段一直难以精确地确定基因型。在高度多态性hla基因座处这种难度增加。用以前的hla分型方法使用hapmaprnaseq数据库来预测四位数hla类型一直是不精确的(图3)。例如,seq2hla方法不适合用于解析四位数hla类型,其具有32%的低精确度(boegel等人,genomemed.(《基因组医学》)4:102(2013))。当将hlaminer应用于该数据集时,仅有可能仅以比对模式执行该方法,因为其contig组装模式由于短的读段长度而无法起作用。得到的精确度仅为39.8%(图3)。hlaforest达到84.2%的较高但仍次优的预测精确度(图3)。
利用相同的hapmaprnaseq数据集,使用实例1的phlat方法在i类基因座处正确推测出96.2%的四位数hla类型,对于组合的i类和ii类基因座二者总体上为92.3%(图3)。phlat还精确地预测了纯合子识别(call)。在四位数分辨率的45个纯合基因座(90个等位基因)中,仅6个被错误地分型为杂合的(总共7个假等位基因)。大部分错误分型的等位基因在两位数分辨率下是精确的,并且与真等位基因仅相差一个或两个核苷酸。
此外,phlat比以前的方法更精确地预测了两位数hla类型。phlat在564个两位数等位基因中仅错误预测5个(精确度为99.1%),而对于该数据集,以前的hla预测方法的两位数精确度不高于97.3%(图3)。
phlat还提供了排除十分罕见的hla等位基因的选择,该十分罕见的hla等位基因在allelefrequencynet不具有任何关于种群频率的记录。在具有该选择的情况下,对最有可能的hla类型的搜索减少至hla-a(526个)、hla-b(674个)、hla-c(373个)、hla-dqa1(33个)、hla-dqb1(81个)、hla-drb1(407个)基因座处的2094个等位基因。当排除罕见等位基因时在这些条件下使用phlat导致四位数分辨率下的精确度为93.0%,与包括罕见等位基因在内时的精确度相当(92.3%,见上文)。
实例3:phlat使用较低覆盖度测序数据精确地确定hla类型
hapmap全外显子测序(wxs)数据集和伴随的i类四位数hla类型收集自祖先来自北欧和西欧、日本和尼日利亚的犹他州居民。wxs数据经由研究登录号srp004078、srr004076和srr004074从公用数据库获得,hla基因型取自warren等人,genomemed.(《基因组医学》)4:95(2012)以及abecasis等人,nature(《自然》)467:1061-1073(2010)。测序通过双端101bp读段来处理,hla基因座的cds区上的中值覆盖度为大约60倍(还可参加“结果”)。
phlat和其他程序用来自ceu、jpt和yri群体的15个hapmap个体的2×101bp全外显子测序(wxs)数据来评价。读段长度较于hapmaprnaseq数据的读段长度要长得多。然而,测序深度降低。对于感兴趣的hla基因座,映射后的深度为大约60倍,而hapmaprnaseq数据集具有大约330倍。尽管该覆盖度倍数对于一般的基因型分型而言可认为是足够的,但对于高度多态性hla基因座的精确分型而言可能存在挑战。
使用wxs数据集的多种hla分型方法的性能在图3中提供。将hlaminer的组装模式应用于该数据集,因为其给出好于比对模式的结果,大概是因为contig在与等位基因的序列比对中比独立的读段更有用,并且对覆盖度的依赖性更低。在四位数分辨率下,hlaminer的精确度为53.3%。还采用默认参数在本地对同一数据集执行hlaforest,得到的精确度为45.6%。与hapmaprnaseq数据集相比,使用wxs数据集时hlaforest的性能更差,尽管wxs数据具有长得多的读段。
当将实例1中描述的phlat方法应用于wxs数据时,其得到的四位数分型精确度为93.3%。此外,phlat给出的两位数精确度为95.6%,高于seq2hla(93.3%,p值没有阈值)并且远优于hlaminer(78.9%)和hlaforest(81.1%)。
实例4:将phlat应用于靶向扩增子测序数据
将实例1中描述的phlat方法应用于靶向扩增子测序数据。该数据通过利用pcr扩增法扩增五个人细胞系中的i类hla-a和hla-b基因座而产生(图4)。简单而言,在第一轮pcr中,产生hla-a和hla-b基因座处的外显子2和3的扩增子(引物序列在图5中提供)并同时添加illumina测序接头。将四种扩增子以1:1:1:1比率汇集并利用第二轮pcr产生条形码。最后,在illuminamiseq(加利福尼亚州的illumina公司(illuminainc.ca))上通过2×250个循环的多重双端测序对汇集的五份样品进行测序。通过miseqreporter软件获得这五份样品的经去重处理(de-multiplexed)的fastq文件。
还通过如下sanger测序对这五份样品的hla-a和hla-b基因座进行基因型分型。通过
实例1的phlat方法使用bowtie2比对工具,bowtie2比对工具能够同时处理短读段和长读段。在5份样品的双端250bp扩增子测序数据集上测试phlat。对于hla-a和hla-b基因座处的总共20个实验验证的等位基因,phlat在两位数和四位数分辨率下均以100%的精确度预测了hla类型(图3)。hlaminer例外,以前公开的方法不能利用该测序数据预测hla类型。在运行hlaminer的组装模式后,对于四位数和两位数分辨率分别获得了50%和95%的精确度。
实例5:错误分型的等位基因的表征
phlat中错误分型的四位数等位基因收集自hapmaprnaseq、1000genomewxs和hapmapwxs数据集,并根据等位基因类型汇总(图6a)。研究了某些等位基因类型是否被富集,并且如果富集的话,则是否是因为该算法或其他原因引入它们。在hla-a、b、c和drb1基因座处,几乎所有的等位基因均具有有限的样本大小(≤10的总发生率)和错误分型事件(≤2)。因而,对于等位基因类型并没有明显的富集。
在hla-dqa1和hla-dqb1基因座,一些特定的等位基因在观察到的预测误差中占优。如图6a中所示,hla-dqa1处的总共二十个错误预测中,十个hladqa1*03:01等位基因被分型为hla-dqa1*03:03,六个hla-dqa1*05:01等位基因被错误地当成hladqa1*05:05。在hla-dqb1基因座处,五个hla-dqb1*02:01等位基因被认为是hla-dqb1*02:02。这些错误占hla-dqa1和hla-dqb1基因座处的全部错误预测的>80%。这些等位基因还在本研究中展现出低的预测精确度(61.5%-73.7%)。尽管真实和预测的等位基因在序列上是高度同源的(<=3snp),但下面的一些观察结果表明,这些误差可能不是随机的。
其他的算法即hlaforest和hlaminer展现出将phlat错误识别的相同样品中的dqa1*03:01错误分型为dqa1*03:03的类似倾向。在七份样品中,hlaforest产生与phlat相同的错误。来自hlaminer的输出dqa1*03:01p是将dqa1*03:01、dqa1*03:03和一些其他等位基因当成一组的p-名称(p-designation)注解。在没有p名称的情况下再次运行hlaminer表明,在phlat错误分型的所有样品中dqa1*03:03是最确信的预测。因为在执行不同比对工具(如,phlat的bowtie2、hlaforest的bowtie以及hlaminer的bwa)的算法中出现相同的错误,所以该错误不是由具体的比对引擎引起的。的确,在任何受影响的样品中,在phlat中将比对工具换成bwa并不改变输出。这些结果表明,该问题可能不是由于算法中的计算策略或比较工具选择所致。
在所有情形中,足够数量的读段支持dqa1*03:03的推断。图6b示出了发生错误分型的一份代表性样品(受试者na12156)中区分dqa1*03:01等位基因和dqa1*03:03等位基因的单snp位点(chr6:32609965,对于dqa1*03:03是碱基a,对于dqa1*03:01是碱基c)周围的读段映射细节。该样品中的第二个等位基因为dqa1*02:01,在该位置其序列为c。这些读段已经被送入phlat流程并被用于hla预测。在样品na12156中,约一半的碱基为a,从而导致杂合基因型ac。因而,在已知这些数据的情况下,推断出dqa1*03:03等位基因还有dqa1*02:01等位基因是有说服力的。对于具有dqa1*03:03预测的所有其他样品也有类似的观察结果。这表明,该误差可能不是简单地由于数据中的随机噪声所致
有可能是,支持所述备选等位基因的读段源自基因组中的别的地方。用带有来自hla-dqa1*03:03等位基因的snp位点(chr6:32609965)的135核苷酸片段(chr6:32609874-32610008)进行blast查询,返回位于hladqa2基因的外显子3处的最靠前全长命中序列。除了该区域内两个等位基因之间仅有的snp位点外,不存在其他错配(图6c)。由于对hladqa2的等位基因了解有限,imgt数据库不包括任何hladqa2条目。因此,所有以前的算法在它们的映射参考序列中不具有hladqa2序列。phlat将该参考序列扩展到全基因组。然而其仅包括hg19基因组中使用的一个特定hla-dqa2等位基因的序列,从而也未能完全地捕集其多态性。鉴于高的序列同源性以及缺少hla-dqa2的完整等位基因参考序列,错误地将hla-dqa2基因的读段比对至hla-dqa1基因的可能性不可忽略。事实上,在chr6:32713784处存在hla-dqa2基因的常见的c到a错义snp(rs62619945,约4%的次要等位基因频率,图6c),该位点为dqa1*03:03等位基因snp在序列比对中的匹配位点。因而,如果受试者恰好携带具有rs62619945snp的特定hla-dqa2等位基因,则所导致的读段可能会被错误地当成来自hla-dqa1*03:03等位基因。
对于其他两个经常被错误分型的等位基因(hla-dqa1*05:01和hla-dqbi*02:01)存在类似的观察结果。在五份样品中,phlat、hlaminer和hlaforest(无p名称)全部错误地分别将它们鉴定为hla-dqa1*05:05和hla-dqb1*02:02。在chr6:32605266、chr6:32610002和chr6:32610445处存在三个驱动dqa1*05:05识别(call)的snp。每个snp具有大量的支持dqa1*05:05等位基因的映射读段。此外,每个snp位于与hla-dqa2基因同源的外显子片段(取自dqa1*05:05等位基因的序列)内。这些片段长度为72-116个核苷酸并且在2-4个染色体位置处与hla-dqa2序列(hg19基因组)不同。hla-dqa2基因中的所有位置均具有dbsnp记录,其中备选碱基匹配dqa1*05:05等位基因中的序列。因而,对于这些区域,有可能混淆来自hla-dqa2和hla-dqa1基因座的读段。类似地,较于hla-dqb1*02:01等位基因(chr6:32629905),该snp偏好hladqb1*02:02等位基因。其处于hla-dqb1基因和hla-dqb2基因之间的91个核苷酸的同源区域内。对hla-dqb2等位基因的研究不充分并且也未记录在imgt数据库内。
总体考虑上述结果,我们推论,将来自次要hla-dqa2基因座和dqb2基因座的读段分别错误地比对至它们的同源主要hla-dqa1基因座和dqb1基因座,可能导致了错误分型的hla-dqa1等位基因和dqb1等位基因的频率不同寻常地高。这种局限性不依赖于算法。将hla-dqa2和dqb2的等位基因序列并入映射参考序列中,将有可能减轻该问题。当使用具有100bp或更长的双端读段的数据时,不太担心错误分型的等位基因的问题,因为本文讨论的同源区域有大约100个核苷酸。长的测序读段可扩展至同源性较低的周边区域来降低错误比对。phlat或其他现有的算法的使用者可通过sanger或靶向扩增子测序来验证hladqa1*03:03、hla-dqa1*05:05和hla-dqb1*02:02等位基因类型。
实例6:影响hla推断的精确度的因素
将来自上述数据集的phlathla预测结果进行汇编,以系统地研究测序参数是如何影响hla推断的精确度的。基准数据集在各种读段长度(37bp-250bp)和深度(从<60倍至>1000倍)以及不同测序方案(双端测序或单端测序)方面提供了测试案例。
图2示出了来自以下三个数据集的结果:hapmaprnaseq、1000genomewxs和hapmapwxs。hapmaprnaseq数据集和hapmapwxs数据集在实例2和3中进行了描述。
对于每个数据集,根据样品的在hla基因座处的映射后覆盖度倍数(x-轴)来将样品进行分仓。符号的y坐标表示样品每个仓内的平均精确度(四位数分辨率下),误差棒指示方差。对于每个双端测序数据集(实心符号),还在单端假设(空心符号)下通过忽略读段之间的成对关系来处理样品。通过平滑线条示出符号的趋势,该平滑线条通过样条内插得到。
如图2中所示,phlat方法的精确度与覆盖度倍数正相关。增加覆盖度倍数时精确度的上升趋势不仅在单独的数据集内出现而且还在它们之间出现。例如,覆盖度系统地高于hapmapwxs样品的1000genomewxs样品一致性地展现出更高的精确度,尽管这两个数据集的其他测序参数相似。这种依赖关系可能有助于估计用于phlat的经验覆盖度阈值以便达到最佳预测。为了在双端测序中实现不低于90%的精确度(水平的虚线,图2),可能要应用30倍-50倍覆盖度,对于低于100bp的读段长度,覆盖度要大于100倍。
当忽略成对约束并且将读段视作单端测序读段时,对于所有数据集均观察到预测精确度有不可忽略的系统性降低。在图2中,分别对于双端测序读段(2×101bp,底部分图,实心圆圈)和单端测序读段(1×101bp,底部分图,空心圆圈),hapmapwxs数据的精确度从>90%降到~85%。在hapmaprnaseq数据中降低更明显:从90-95%(2×37bp,顶部分图,实心圆圈)降到70-90%(1×37bp,顶部分图,空心圆圈)。这些观察结果凸显了双端测序对hla类型推断的重要性。成对读段的优势源于实际上加倍的读段长度,这降低了映射的模糊性。此外,长的末端到末端的跨度(通常数百个碱基)连接了相对远离的snp,从而使得phlat能利用长的范围内来自snp对的相位信息。
序列表
<110>bai,yu
fury,wen
<120>高分辨率等位基因鉴定
<130>37595.0008u2
<150>pct/ib2014/002843
<151>2014-10-14
<150>61/891,193
<151>2013-10-15
<160>25
<170>patentin版本3.5
<210>1
<211>19
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>1
crggtctcagccactsctc19
<210>2
<211>18
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>2
ctcggacccggagactgt18
<210>3
<211>19
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>3
ctygggggacygggctgac19
<210>4
<211>21
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>4
cccaattgtctcccctccttg21
<210>5
<211>18
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>5
ggsagggaaatggcctct18
<210>6
<211>19
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>6
ggatggggagtcgtgacct19
<210>7
<211>19
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>7
gcgtttacccggtttcatt19
<210>8
<211>20
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>8
cggcgacctataggagatgg20
<210>9
<211>45
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<220>
<221>修饰的碱基
<222>(23)..(26)
<223>n可以是a、c、t、g、未知或其他
<400>9
ctacacgacgctcttccgatctnnnncrggtctcagccactsctc45
<210>10
<211>40
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>10
cagacgtgtgctcttccgatctctcggacccggagactgt40
<210>11
<211>45
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<220>
<221>修饰的碱基
<222>(23)..(26)
<223>n可以是a、c、t、g、未知或其他
<400>11
ctacacgacgctcttccgatctnnnnctygggggacygggctgac45
<210>12
<211>43
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>12
cagacgtgtgctcttccgatctcccaattgtctcccctccttg43
<210>13
<211>44
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<220>
<221>修饰的碱基
<222>(23)..(26)
<223>n可以是a、c、t、g、未知或其他
<400>13
ctacacgacgctcttccgatctnnnnggsagggaaatggcctct44
<210>14
<211>41
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>14
cagacgtgtgctcttccgatctggatggggagtcgtgacct41
<210>15
<211>45
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<220>
<221>修饰的碱基
<222>(23)..(26)
<223>n可以是a、c、t、g、未知或其他
<400>15
ctacacgacgctcttccgatctnnnngcgtttacccggtttcatt45
<210>16
<211>42
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>16
cagacgtgtgctcttccgatctcggcgacctataggagatgg42
<210>17
<211>58
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>17
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58
<210>18
<211>66
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>18
caagcagaagacggcatacgagatgattagccgtgactggagttcagacgtgtgctcttc60
cgatct66
<210>19
<211>66
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>19
caagcagaagacggcatacgagattgcgacatgtgactggagttcagacgtgtgctcttc60
cgatct66
<210>20
<211>66
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>20
caagcagaagacggcatacgagatcagcgttagtgactggagttcagacgtgtgctcttc60
cgatct66
<210>21
<211>66
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>21
caagcagaagacggcatacgagattccgtaaggtgactggagttcagacgtgtgctcttc60
cgatct66
<210>22
<211>66
<212>dna
<213>人工序列
<220>
<223>合成的构建体;引物
<400>22
caagcagaagacggcatacgagatatggtaccgtgactggagttcagacgtgtgctcttc60
cgatct66
<210>23
<211>35
<212>dna
<213>智人(homosapiens)
<400>23
accttcctcccttctgctgatgagatttatgactg35
<210>24
<211>18
<212>dna
<213>智人(homosapiens)
<400>24
tcagtcacagaaggtgtt18
<210>25
<211>87
<212>dna
<213>智人(homosapiens)
<400>25
cattccttcttcaagatcagttacctcaccttcctcccttctgctgatgagatttatgac60
tgcaaggtggagcactggggcctggac87
- 还没有人留言评论。精彩留言会获得点赞!