一种RNA编码潜能的预测方法与流程
本发明属于基因注释领域,更具体地,涉及一种rna编码潜能的预测方法。
背景技术:
近几年来,下一代测序技术产生了成千上万新的转录本,于是快速且准确地区分编码rnas和非编码rnas(ncrnas)成为分析这些数据的关键。在生物体中,ncrna虽然不能编码蛋白质但是也具有重要的生物功能,比如基因调控、基因沉默、rna修饰和加工。
在编码潜能的预测领域,已经公开了一种使用无比对逻辑回归模型的编码潜能评估工具cpat。其使用4个序列特征:开放阅读框的长度、开放阅读框的覆盖率、fickett打分和六聚体打分。此预测领域中,还公开了cpc2,其也只是使用4个序列特征:开放阅读框的长度、fickett分数,开放阅读框的完整性和等电点。另外一种工具plek,使用改进的k-mer策略预测长链非编码rna和编码rna。虽然这些工具可以很好的区分长的编码rna和ncrna,但是对于sorf的编码潜能的预测精度较低,故在预测sorf上仍存在很大不足。
目前,越来越多的sorf的数据被发现,然而具有意义和功能的sorfs的数目比较少。2010年,sorffinder被提出,它是一种专门为预测sorf设计的程序,它只使用六聚体特征来预测sorf的编码。然而,只使用一个特征得到的预测结果会有很高的假阳性率。所以,在sorf预测方面仍然是一个悬而未决的问题。故本发明提出一种rna编码潜能的预测方法cppred,不仅能够很好的预测长的rna序列,而且对于短的rna序列的预测也有较高的准确性。
技术实现要素:
针对现有技术的以上缺陷或改进需求,本发明提供了一种rna编码潜能的预测方法,该方法(命名为cppred)通过整合多个序列特征,特别是本发明使用ctd来描述rna的全局分布;然后,以候选特征之间的冗余度和相关性作为标准,并结合递增特征选择方法,从中选取最佳特征集合作为特征向量;通过支持向量机(svm)建立预测模型;最后根据待预测的rna序列的特征向量,获取预测结果。本发明提供的预测方法在预测长的rna序列和当前已有方法结果相当(准确度达到90%以上),然而在短的rna序列预测上,该方法明显优于当前已有的方法。由此解决现有技术的sorf的编码潜能的预测方法和工具存在的预测准确度不高以及存在过拟合风险的技术问题。
为实现上述目的,按照本发明的一个方面,提供了一种rna编码潜能的预测方法,包括如下步骤:
(1)训练集中的rna样本候选特征集合的获取:所述候选特征集合包括开放阅读框的长度、开放阅读框的覆盖率、六聚体分数、fickett分数、开放阅读框的完整性、多肽的等电点、多肽的亲水性、多肽的不稳定性以及ctd编码特征;
(2)最佳特征集合的获取:根据步骤(1)所述候选特征集合中各特征之间的相关性和冗余度选择方法获取最佳特征集合;
(3)使用步骤(2)获得的最佳特征集合,对训练集中所有的rna样本采用机器学习方法进行训练,获取rna编码潜能预测模型;
(4)将待预测rna序列对应的最佳特征集合代入到步骤(3)中所述的预测模型,得到待预测rna序列的预测结果。
优选地,所述ctd编码特征表示全局转录本序列描述符,其中:
第一个描述符c用于描述转录本序列中每个核苷酸的百分比组成;
第二个描述符t用于描述相邻位置之间四个核苷酸转换的频率百分比;
第三个描述符d用于描述每个核苷酸转录序列上的五个相对位置,分别为0、25%、50%、75%和100%,其中0代表第一个相对位置,100%代表最后一个相对位置。
优选地,步骤(2)具体为:采用最大相关最小冗余方法对所述候选特征集合中的特征进行排序,结合递增特征选择方法使用交叉验证方法进行训练和测试,获取样本的评估指标σ,选取σ最大时的特征集合作为最佳特征集合。
优选地,所述评估指标σ为马修相关系数。
优选地,步骤(2)具体为:对步骤(1)所述候选特征集合中各特征采用主成分分析pca选择方法获取最佳特征集合。
优选地,步骤(3)获得rna编码潜能预测模型以后,将测试集中的rna序列对应的最佳特征集合代入到所述的预测模型,得到测试集中rna序列的预测结果,以验证所述预测模型的准确性。
优选地,所述训练集用于建模,所述测试集用于检测模型的准确性,使用时对所述训练集内部进行去冗余操作,以减少过拟合风险;同时训练集和测试集之间也进行去冗余操作。
优选地,步骤(3)所述机器学习方法为支持向量机法。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果:
(1)本发明的rna编码潜能预测的方法cppred不依赖于rna的长度,通过引入ctd编码的特征,cppred既能区分长的编码rnas和ncrnas,又能很好的区分短的编码rnas和ncrnas;在短序列预测上更有优势,相较于现有技术的短序列预测方法准确度高。
(2)本发明首次使用了ctd编码的转录本特征来预测真核生物的rna的编码潜能,再结合现有工具的特征开发的一种新的预测编码潜能的工具。该工具大大降低了物种依赖性,具有良好的物种普适性。
(3)本发明rna编码潜能的预测方法通过对选择的训练集和测试集进行去冗余操作,并通过精心选择特定的候选特征集合,再对候选特征进行排序,获得最佳特征集合,利用最佳特征集合进行模型的构建以及待测序列的预测,本发明rna编码潜能预测工具没有过度拟合,也不存在过度拟合的风险。
(4)在确定最终的预测模型时,以马修相关系数作为评估指标,比通过准确度评估更具说服力,建立的预测模型更为优越。
附图说明
图1为本发明rna编码预测方法流程图;
图2为本发明数据集的构建流程图;
图3为本发明中ctd编码的特征的示例;
图4为本发明中候选特征集合中特征的排名示意图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
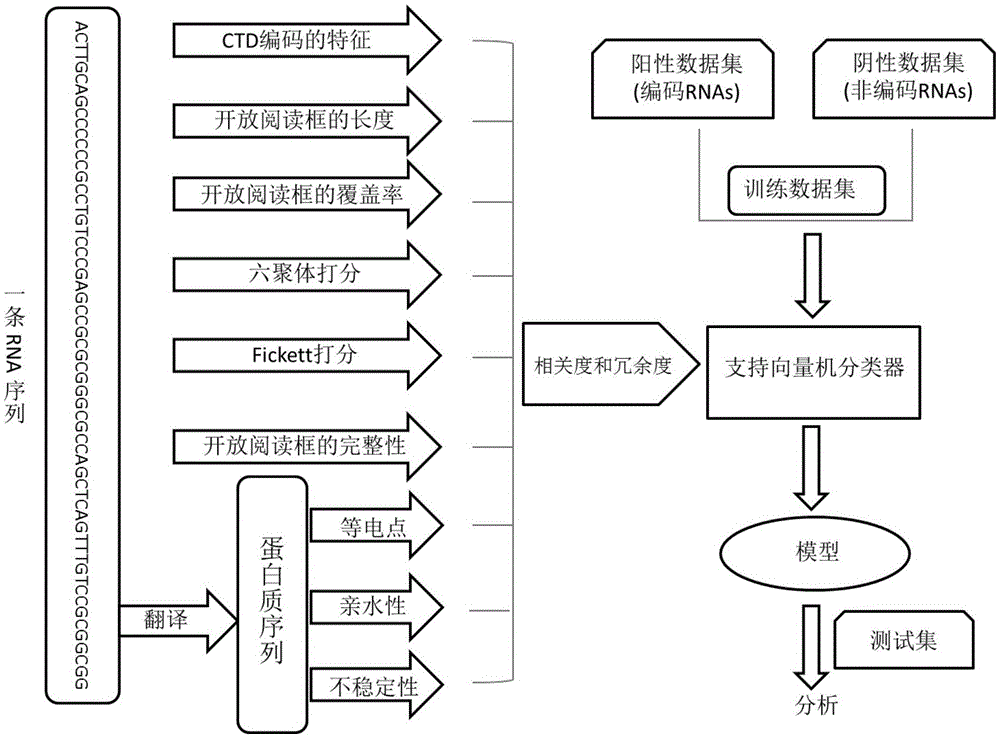
本发明公开了一种rna编码潜能的预测工具cppred,如图1所示,其原理步骤包括:
(1)训练集中的rna样本候选特征集合的获取;选取来自rna序列和蛋白质序列的多个特征,其包含之前开发的cpat(wangetal.,2013,cpat:coding-potentialassessmenttoolusinganalignment-freelogisticregressionmodel.nucleicacidsres)和cpc2(kangetal.,2017,cpc2:afastandaccuratecodingpotentialcalculatorbasedonsequenceintrinsicfeatures.nucleicacidsres)软件的特征。本发明所述候选特征集合包括开放阅读框的长度、开放阅读框的覆盖率、六聚体分数、fickett分数(通过核苷酸组成和密码子使用偏差的组合效应来计算得到)、开放阅读框的完整性、多肽的等电点、多肽的亲水性、多肽的不稳定性以及ctd编码特征;本发明特别在候选特征集合中增加了ctd编码的特征,其是首次应用到真核生物预测rna编码潜能的特征。
本发明数据集包括测试集和训练集,训练集用于建模,测试集用于检测模型的准确性。本发明选取训练集或测试集时,训练集内部进行去冗余操作,以减少过拟合风险,得到更普适的模型。同时训练集和测试集之间要也进行去冗余,可避免训练过的数据用于测试,失去测试的意义。然而,之前预测编码潜能的工具在构建数据的时候却没有去冗余操作,这样可能存在过拟合的风险。
一些实施例中,如图2所示,将refseq数据库中所有人类的mrna数据作为阳性样本,随机选取2/3作为训练集,剩余的1/3作为测试集。将ensembl数据库中所有人类的非编码rna数据作为阴性样本,首先,删除没有注释来源的数据;然后,在剩余的数据中随机选取2/3作为训练集,剩余的1/3作为测试集。训练集内部使用cd-hit方法按照序列一致性阈值大于或者等于99%去冗余。同时,为了确保训练集和测试集之间也是非冗余的,使用cd-hit方法对训练集和测试集按照序列一致性阈值大于或等于80%进行去冗余操作。这样,得到人类测试集,其包括8557条编码rna序列和8241条非编码rna序列。随后,从人类测试中的编码rna中提取出长度小于303个核苷酸的orf片段的rna序列。同时,将来自ncrnas的相当数量的相当长的ncrna随机筛选出来。可以得到,短序列的测试集,包括641条编码的rna序列和641条非编码的rna序列。
ctd是预测蛋白质的折叠时被提出的,其是描述全局蛋白质序列的描述符。本发明中,ctd用于描述全局转录本序列的描述符。
rna是含有四种核苷酸a,t,g和c的序列。第一个指数c描述了转录本序列中每个核苷酸的百分比组成。第二描述符t描述了相邻位置之间四个核苷酸转换的百分比频率。随后,计算每个核苷酸沿着转录本序列的五个相对位置,其中0(第一个),25%,50%,75%和100%(最后一个),以描述最后的描述符d。
ctd编码的特征的详细过程如图3所示,以一条40个碱基的rna序列为例,该序列包括4个腺嘌呤(as),4个胸腺嘧啶(ts),12个鸟嘌呤(gs)和20个胞嘧啶(cs)。对于as,第一描述符c是4/40=10.0%,对于bs,4/40=10.0%,对于gs,12/40=30.0%,对于cs,20/40=50%。对于第二描述符t,在a和t之间存在零转变,a和g之间存在四个转变,a和c之间有三个转变,t和g之间有三个转变,t和c之间有三个转变,g和c之间有四个转变。因此,这些转变的频率分别为0/39=0.0,4/39=0.103,3/39=0.077,3/39=0.077,3/39=0.077和16/39=0.410。第一个,25%,50%,75%和100%的as分别位于1,1,7,25和40个残基上,则as的d描述符是1/40=0.025,1/40=0.025,7/40=0.175,25/40=0.625和40/40=1.0。同样,ts的d描述符是0.075,0.075,0.10,0.450和0.50,对于gs是0.125,0.375,0.650,0.825和0.925,对于cs是0.050,0.275,0.425,0.70和0.975。如图4所示,由ctd编码的特征t2和c0在预测编码潜能中发挥着重要的作用。
(2)最佳特征集合的获取:根据步骤(1)所述候选特征集合中各特征之间的相关性和冗余度选择方法获取最佳特征集合。
一些实施例中,步骤(2)具体为:采用最大相关最小冗余方法(mrmr方法)对所述候选特征集合中的特征进行排序,结合递增特征选择方法使用交叉验证方法进行训练和测试,获取样本的评估指标σ,选取σ最大时的特征集合作为最佳特征集合。评估指标σ可以为马修相关系数,也可以为其他常规评估指标,但优选马修相关系数。马修相关系数为一个综合的评估指标,以马修相关系数作为评估指标,比通过准确度评估更具说服力,建立的预测模型更为优越。
递增特征选择方法,一些实施例中具体为:首先选择使用mrmr方法排名第一的特征进行训练建立模型,计算其10倍交叉验证的评估性能,然后选用排名在前2个的特征进行建模,也计算10倍交叉验证的性能,以此类推,每增加一个排名在前面的特征,就得到一个模型,直至特征全部添加完毕。
一些实施例中,采用最大相关最小冗余方法(mrmr方法)对训练样本集的候选特征集合中的特征进行排序后,结合递增特征选择方法使用十倍交叉验证方法进行训练和测试,获取排序后38个特征递增叠加的马修相关系数mcc1,mcc2…mcc38,其分别表示1个,2个…38个特征递增叠加后对应的马修相关系数。选取mcc最大时对应的特征集合作为最佳特征集合。将mcc1,mcc2…mcc38的值作为纵坐标,将特征个数作为横坐标,作图如图4所示,可以看出,采用上述方法排序后,38个特征中前七个特征,包括orf的完整度、orf的覆盖率、多肽的稳定性、ctd编码的特征(t2,c0)、多肽的等电点和orf的长度递增叠加马修相关系数增长速率较大,而后31个特征递增叠加马修相关系数增长速率平缓,说明在候选特征集合中,orf的完整度、orf的覆盖率、多肽的稳定性、ctd编码的特征(t2,c0)、多肽的等电点和orf的长度是预测编码潜能中的重要特征,这样的组合特征对于编码潜能的预测具有重要作用。进而也说明本发明选择的ctd编码特征t2和c0在rna编码潜能预测中的作用不容忽视。
如图4所示,当特征个数是37的时候,mcc值最大(mcc=0.953),故选择前37个特征作为最佳特征集合。
对步骤(1)所述候选特征集合中各特征也可采用主成分分析pca选择方法获取最佳特征集合,其能够获得和上述方法相同的最佳特征集合的特征数目。
(3)使用步骤(2)获得的最佳特征集合,对训练集中所有的rna样本采用机器学习方法进行训练,获取rna编码潜能预测模型;一些实施例中机器学习方法为支持向量机法(svm)。
(4)将测试集中的rna序列对应的最佳特征集合代入到所述的预测模型,得到测试集中rna序列的预测结果,以验证所述预测模型的准确性。
(4)将待预测rna序列对应的最佳特征集合代入到步骤(3)中所述的预测模型,得到待预测rna序列的预测结果。svm方法默认阈值为0.5,模型输出值大于或等于该阈值,表明该待测rna序列为可编码序列,否则为非编码序列。
方法评估:
将测试集中的rna序列对应的最佳特征集合代入到上述步骤中所建的预测模型,得到待预测样本的预测结果,本发明使用多种评估指标,分别包括灵敏度(sn)、特异性(sp)、精确度(pre)、准确度(acc)、f-measure、马修相关系数(mcc)、受试者操作特性曲线下的面积(auc,areaunderthereceiveroperationcharacteristiccurve),具体如下:
其中,tp为真阳性,指阳性数据中被正确预测为阳性的个数;fn为假阴性,指阳性数据中被错误地预测为阴性的个数;tn为真阴性,指阴性数据中被正确地预测为阴性的个数;fp为假阳性,指阴性数据中被错误地预测为阳性的个数。
从mcc的定义来看,它是预测结果的一种综合评估。对于auc,其是以灵敏度为纵坐标,特异性为横坐标绘制的曲线与x轴围成的面积。它考虑在不同阈值下对应的sn和sp值,故auc也是一种对预测结果进行综合评估的指标。
实施例
本发明使用cppred测试了人类,小鼠,斑马鱼和酿酒酵母的数据,并与现有的cpat、cpc2、plek、sorffinder工具测试结果进行比较。
在人类测试集(包括长序列和短序列)和人类sorf的测试集上,不同预测工具预测性能比较结果见表1和表2。从表1和表2可以看出,不论是人类测试集还是人类sorf的测试集,cppred均优于cpat和cpc2,然而略差于plek。这是因为plek的训练集和人类测试集之间有冗余。
表1:cppred与cpat、cpc2、plek人类的测试集上的比较
表2:cppred与cpat、cpc2、plek、sorffinder在人类sorf的测试集上的比较
在小鼠测试集和小鼠sorf的测试集上,测试结果见表3和表4。从表3和表4可以看出,cppred都优于其他几种方法(表格3和4)。
表3:cppred与cpat、cpc2、plek在小鼠测试集上的比较
表4:cppred与cpat、cpc2、plek、sorffinder在小鼠sorf测试集上的比较
在斑马鱼测试集和斑马鱼sorf的测试集上,测试结果见表5和表6。从表5和表6可以看出,cppred都优于其他几种方法。
表5:cppred与cpat、cpc2、plek在斑马鱼测试集上的比较
表6:cppred与cpat、cpc2、plek、sorffinder在斑马鱼sorf测试集上的比较
在酿酒酵母测试集和酿酒酵母sorf的测试集上,测试结果见表7和表8。从表7和表8可以看出,cppred都优于其他几种方法。
表7:cppred与cpat、cpc2、plek在酿酒酵母测试集上的比较
表8:cppred与cpat、cpc2、plek、sorffinder在酿酒酵母测试集上的比较
另外,作为对比,使用ctd特征训练的模型(octd-model)和只使用开放阅读框的长度,开放阅读框的覆盖率,六聚体分数,fickett分数,开放阅读框的完整性,多肽的等电点,多肽的亲水性,多肽的不稳定性,即非ctd编码的特征训练的模型(nctd-model),然后在人类sorf数据上进行测试。这里cppred在人类sorf数据的性能也列在表9中。结果表明ctd编码的特征更有利于短的rna序列的预测。
表9:在人类sorf的测试集上octd-model,nctd-model和cppred的性能
从上述表1至表8可以看出,本发明的cppred在人类,小鼠,斑马鱼和酿酒酵母测试集上,具有高的准确性,相比于cpat,cpc2和plek工具的准确性有微弱的提高,然而,本发明的cppred在这些物种的短的rna序列上具有特别的优势,比之前开发的工具有一个比较大的提升。这可能是因为,本发明使用了ctd编码的特征,而ctd编码的特征与rna的二级结构具有相关性,rna的二级结构在rna编码中起着重要作用,故cppred捕获了二级结构的特征,从而在预测性能上更具有特别的优势。
本发明中cppred是在人类数据上进行的训练,在多个物种的测试集上进行了测试,如表格1-8结果显示,cppred预测准确性相对来说都比较高,故该工具大大降低了物种依赖性,具有良好的物种普适性。这可能是在训练集构建的时候,进行了去冗余操作,避免了过拟合的情况,得到的模型不具有某种偏好性,得到了更普遍适用性的模型。
本发明为了进一步评估cppred,于是对最近新发现的人类编码rna进行了测试,从2017年11月27日到2018年4月3日,refseq数据库中获得了74条新的人类编码rna序列,其中包括5条短的rna序列。cppred成功预测了74条新的人类编码rna序列中的67条序列,成功预测了新的人类短的rna序列中的4条。由此可以看出,cppred具有较强的预测能力。
- 还没有人留言评论。精彩留言会获得点赞!